Determination of saturated aliphatic hydrocarbons in vegetable oils
R.B. Gómez-Coca*, R. Cert, M.C. Pérez-Camino and W. Moreda
Department of Characterization and Quality of Lipids, Instituto de la Grasa – CSIC-Campus of Universidad Pablo de Olavide, Building 46, Ctra. Utrera km 1, 41013 Seville, Spain
*Corresponding author: raquel.coca@ig.csic.es
| |
SUMMARY
The aim of this work is to inform about the development of a simple and reliable off-line method for the determination of
saturated hydrocarbons (SH) in vegetable oils. SH can be used as markers for fuel or for mineral oil contamination in edible
oils and fats. The method consists of the isolation of the fraction by LC on deactivated silver-silica gel and subsequent
on-column GC-FID analysis. This stationary phase was prepared avoiding any kind of activation. The method was developed and
validated through the participation in both a proficiency test organized by the Joint Research Centre of the European Commission,
and a collaborative trial carried out with trained Spanish laboratories according to the standard ISO 5725. Results showed
acceptable repeatability and reproducibility values, and Horrat index, being this protocol in use with satisfactory results
ever since. The method’s LOQ is 15 mg·kg–1 and its LOD 5 mg·kg–1, which make it suitable to quantify the 50 mg·kg–1 limit established by the EU, and to detect mineral oil content within the 10–500 mg·kg–1 range. Although other procedures with lower LOD have been developed throughout the years, the use of just regular laboratory
equipment such as GC-FID makes the proposed method appropriate for application on a routine basis.
|
| |
RESUMEN
Determinación de hidrocarburos alifáticos saturados en aceites vegetales. El objetivo de este trabajo es el de dar cuenta del desarrollo de un método sencillo y fiable para la determinación de hidrocarburos
saturados (HS) en aceites vegetales. Los HS pueden utilizarse como marcadores de contaminación de aceites y grasas comestibles
con fuel-oil y aceites minerales El procedimiento consiste en el aislamiento de la fracción correspondiente por cromatografía
en columna de gel de sílice argentada sin activar y posterior análisis mediante GC (on-column)-FID. El método se desarrolló
y validó mediante la participación en una prueba de competencia organizada por el Joint Research Centre de la Comisión Europea, además de con un ensayo colaborativo llevado a cabo por laboratorios españoles de acuerdo con la
norma ISO 5725. Los resultados mostraron valores de repetibilidad y de reproducibilidad aceptables, así como del índice de
Horrat, por lo que dicho protocolo se está utilizado con resultados muy satisfactorios. El límite de detección (LDD) es de
5 mg·kg–1, y su límite de cuantificación (LDQ) de 15 mg·kg–1, lo que lo hacen muy adecuado para evaluaciones alrededor del límite de 50 mg·kg–1 establecido por la Unión Europea (UE). Asimismo es fiable para determinar el contenido de aceite mineral en el intervalo
entre 10 y 500 mg·kg–1. Si bien se han desarrollado otros procedimientos con menor LDD a lo largo de los años, el uso de equipos habituales de laboratorio
tales como GC-FID ha hecho que el método propuesto sea el de elección para su aplicación en cualquier laboratorio de forma
rutinaria.
|
1. INTRODUCTIONTOP
Crude edible vegetable oils contain various classes of natural hydrocarbons: 50–350 mg·kg–1 of the n-alkane series from C10 to C35, centred at C25-C29, the odd numbered elements being the most abundant; squalene that
is the major hydrocarbon in olive and pumpkin oils (500–12000 mg·kg–1); and low amounts of the n-alkene series and terpenic hydrocarbons (sesquiterpenes in olive oil, kaurene in sunflower oil).
These compounds, when analysed by high-resolution gas chromatography-flame ionization detector (GC-FID), yield a GC profile
constituted by numerous sharp peaks that can be easily quantified (Lanzón et al., 1994).
On the other hand, base oil for manufacturing the lubricating or hydraulic oils used in the food industry, named “white mineral
oil”, is constituted by a complex mixture of branched saturated aliphatic hydrocarbons (mainly iso-isomers) that yield a GC-FID
hump between C20 and C54, known as unresolved complex mixture (UCM). In GC-FID chromatograms obtained from the hydrocarbon
fraction of vegetable oils, mineral oil contamination can be evidenced by the presence of a hump of branched saturated hydrocarbons
(SH) with a series of sharp peaks on the top corresponding to the n-alkanes.
Nevertheless in refined vegetable oils, significant amounts of unsaturated steroidal hydrocarbons coming from dehydration
of sterols are found, and in refined olive oils, isoprenoid alkenes from isomerization and cyclization of squalene and from
dehydration and cyclization of oxidized squalene are found too (Lanzón et al., 1994; Bastic et al., 1978). All these compounds yield a very complex GC-FID profile between C20 and C35 overlapping with the hump due to mineral oil.
Therefore, the determination of mineral oil in vegetable oils requires the isolation of a SH-fraction from the unsaturated
ones.
GC-FID is the technique of choice for the analysis of the SH fraction because it enables the determination of SH naturally
present in fats and oils, and the hump of branched paraffins from mineral oils. The use of silica gel high performance liquid
chromatography (HPLC) as a first step to separate the paraffins from the rest of the oil has also been described (Fiselier et al., 2009a). Mineral paraffins are determined by the area of the hump, confined by the baseline and an upper contour line defined by
the base line of the sharp (natural) peaks standing on the hump.
Some years ago about 1000 mg·kg–1 of a mixture mainly constituted of saturated aliphatic hydrocarbons ranged from C18 to C40 (centred at C28) was found in
crude sunflower oil coming from Ukraine, and attributed to contamination with mineral oil (Biedermann et al., 2009). To protect consumers the European Commission decided to consider the UCM of SH as marker of mineral oil and established
a legal limit of 50 mg·kg–1 from C10 to C56 (except n-C27, n-C29, and n-C31) in sunflower oils (EC, 2009). At that moment, IUPAC, 1987 and AOCS, 1997 methods were standardized for the control of this contaminant in oils and fats; in the former, the oil is fractioned by thin
layer chromatography (TLC) on silica gel plates and determined by the densitometry of the spot; the latter lies in the weight
of the SH-fraction isolated by column chromatography (CC) on alumina. Both methods suffered from a high detection limit (500–1000
mg·kg–1) and consequently, new methods based on the chromatographic isolation of SH-fraction and posterior analysis by GC-FID were
developed to detect lower amounts of mineral oil.
For the isolation of SH-fraction from the oil CC on alumina, silica gel, and their combinations have been proposed. Studies
on the behaviour of different classes of paraffins (n-, iso-, large carbon-atom number) on alumina showed that the retention
depends on the aluminium oxide activation temperature, mobile phase, column temperature, sensitivity to polar components and
capacity (Fiselier et al., 2009a; Fiselier et al., 2009b; Wagner et al., 2001), and that a partial loss of iso-paraffins may occur (Moret et al., 2011). These results indicate that alumina is not advisable for the quantitative determination of mineral oil.
Silica gel has been usually applied for the isolation of the SH-fraction from the oil, using solid phase extraction (SPE)
(Fiorini et al., 2010), liquid chromatography (LC) (Tan and Kuntom, 1993) and especially on-line HPLC-GC (Tranchida et al., 2011; Biedermann and Grob, 2012). A separation by SPE packed with alumina and silica gel on top, followed by GC-FID analysis with large volume on-column
injection has been described (Fiselier and Grob, 2009). These methods reach sufficient sensitivity but the separation from the unsaturated hydrocarbons is not complete in refined
oils containing significant amounts of sterenes or squalene. To improve resolution and sensitivity, on-line HPLC-HPLC-GC-FID
systems have been proposed where the first column isolates the hydrocarbons from the bulk of the oil and the second one separates
the paraffins (Fiorini et al., 2008; Populin et al., 2004; Neukom et al., 2002). These methods require somehow more sophisticated apparatus that may not be available in many laboratories.
Other approaches to improve the isolation of the SH-fraction from the oil consist of the fractioning of the unsaponifiable
matter on silica gel column, rendering better sensitivity and separation (Lanzón et al., 1994), and the bromination (Wagner et al., 2001; Moret et al., 2003) or epoxidation (Biedermann and Grob, 2012; Biedermann et al., 2009) of the oil to obtain more polar derivatives of the unsaturated compounds that are more strongly retained during LC.
These procedures are solvent- and time consuming, tedious, and prone to contamination.
The use of silver-silica enhances the separation between the SH-fraction and the olefins due to the affinity of the Ag+ ion to double bonds (olefinic and aromatic hydrocarbons) allowing a more reliable fractioning. An off-line method based on
SPE on activated silver-silica gel mixture (1 g) followed by GC-FID has been reported (Moret et al., 2011) for the determination of mineral oil in vegetable oils.
The aim of this work was to inform about the development of a simple and reliable off-line method for the determination of
SH as markers of fuel or mineral oil contamination in edible oils and fats. The criteria were (i) availability of samples
containing large amounts of interfering olefins and (ii) wide concentration range including the legal limit of 50 mg·kg–1 established by the European Commission for Ukrainian sunflower oil (EC, 2009). The method consists of the isolation of the fraction by LC on deactivated silver-silica gel and subsequent on-column GC-FID
analysis. This stationary phase was prepared by a new procedure avoiding the activation used for the isolation of steroidal
hydrocarbons in vegetable oils (Cert and Moreda, 1998).
2. MATERIALS AND METHODSTOP
2.1. SamplesTOP
Samples of sunflower oil coming from Ukraine were obtained from the Spanish Food Safety Agency. Samples of crude and refined
sunflower oils were purchased in the local markets. Refined pomace oil was obtained directly from the producers.
2.2. Material and reagentsTOP
All the reagents were of analytical grade unless otherwise specified. Distilled water, sea sand, and silver nitrate were purchased
from Panreac (Montcada I Reixac, Barcelona, Spain). 3,5-Cholestadiene and the internal standard (IS) n-eicosane (C20) were
from Sigma-Aldrich Co. LLC (St. Louis, Missouri, USA). Diethyl ether, n-heptane, and n-hexane 95% were supplied by Romil Ltd.
(Waterbach, Cambridge, GB). We checked the n-hexane purity by concentrating a mixture of 200 mL of the solvent with 2 mL of
the IS (C20) in a rotary evaporator down to 0.5 mL, and analysing the concentrate by GC with cool on-column injection. Silica
gel 60 for column chromatography, 70–230 mesh, (Merck KGaA, Darmstadt, Germany) was used directly from the container. A silver
nitrate solution (75% w/v) was prepared by dissolving 4.5 g of silver nitrate in 6 mL distilled water. Chromatography columns
(50 cm long × 1.5 cm id) were provided with Teflon stopcocks and were washed with n-hexane before use.
2.3. Method developmentTOP
2.3.1. Separation of the hydrocarbon fractionsTOP
Deactivated 10% silver-silica gel was prepared following the procedure indicated for the separation of steroidal hydrocarbons
(Cert and Moreda, 1998) but decreasing the water proportion down to 13.3% w/v.
For the preparation of 3 chromatography columns, 45 g silica gel were weighted in a 500-mL round-bottomed flask; thereafter
6 mL silver nitrate solution were added drop wise with a Pasteur pipette, shaking then vigorously. The flask was covered with
aluminium foil and put in a rotary evaporator during 30 min at room temperature and atmospheric pressure. To avoid uneven
distribution of the silver nitrate in the silica gel both the condenser and the evaporation flask were set horizontally. Finally
it was let to stand during 12 h before use.
For each column arrangement, 15 g of silver-silica gel were suspended in a beaker in n-hexane, and the slurry was introduced
onto the chromatographic column already containing 40 mL n-hexane. Once the packing settled, a small amount of sea sand (previously
washed with n-hexane) was added (1 cm of column height), and the remaining solvent eluted. In order to avoid bubble formation,
we tapped the column gently with a rubber rod. The column was eluted with 60 mL n-hexane to eliminate impurities of the packing
material, and wrapped in black paper to protect it from the light.
To investigate the behaviour of the different types of hydrocarbons during the chromatographic separation on silver-silica
gel column, refined olive pomace oil spiked with 3,5-cholestadiene was chosen. In a small beaker, 1 g oil was weighed to the
nearest 1 mg, and 1 mL 3,5-cholestadiene standard solution was added. The mixture was transferred to the column with the aid
of a Pasteur pipette and let get in the stationary phase. The beaker was washed with two portions of 1 mL n-hexane that were
also added to the column packing. The column was then eluted with 100 mL n-hexane with a cadence of 15 drops every 10 s approximately,
collecting three separated fractions of 40, 40, and 20 mL, respectively. Subsequently, 120 mL of the n-hexane:diethyl ether
(98:2, v/v) mixture were added, collecting three independent 40 mL fractions. Most of the solvent of each fraction was evaporated
in a rotary evaporator at room temperature under vacuum and until dryness with a nitrogen stream. Each residue was re-dissolved
in 0.5 mL n-heptane and analysed by GC.
2.3.2. Gas chromatography analysis and quantitationTOP
GC analysis of the hydrocarbons was performed using an Agilent 6890N gas chromatograph (Agilent Technologies, Santa Clara,
California) equipped with an Agilent 7683B Automatic Liquid Sampler and FID. Data was acquired with the Agilent ChemStation
for GC system program. Separations were carried out on a high-temperature fused-silica capillary column (5% diphenyl-95% dimethylpolysiloxane:
10 m×0.32 mm id, 0.10 μm film; Sugelabor, Madrid, Spain), 2 μL injection volume, hydrogen carrier gas at 105 kPa and EPC cool
on-column injection. The operating conditions were as follow: injector temperature, 60 °C; detector temperature, 350 °C; oven
programming temperatures, initial 60 °C for 1 min and then rising at 12 °C·min–1 to 350 °C, hold for 4 min. Under these conditions the IS (C20) appeared at a retention time of about 8.5 min (Figure 1).
|
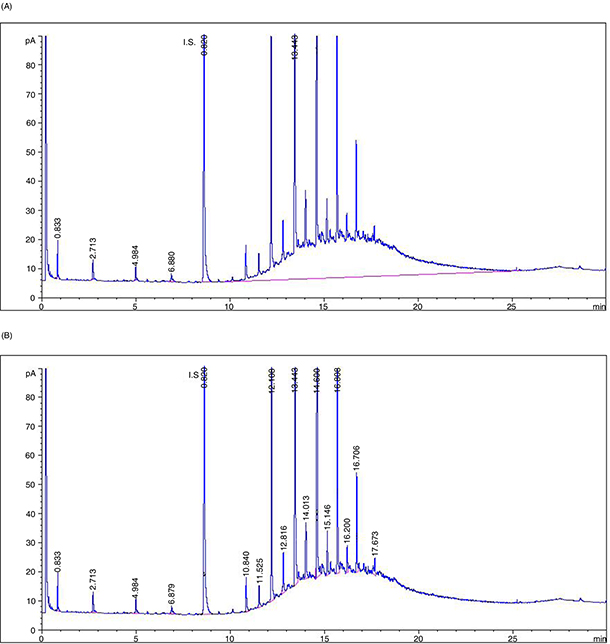 Figure 1. GC profile of the first 40 mL-n-hexane fraction containing SH from refined olive-pomace oil. IS = C20. Oven temperature ramp: 12 °C·min–1. A) Manual baseline traced under hump. B) Baseline traced valley-valley under the natural n-alkane peaks. The main peaks at retention time from 12.1 to 16.7 minutes correspond to the odd series from C23 to C29. Figure 1. GC profile of the first 40 mL-n-hexane fraction containing SH from refined olive-pomace oil. IS = C20. Oven temperature ramp: 12 °C·min–1. A) Manual baseline traced under hump. B) Baseline traced valley-valley under the natural n-alkane peaks. The main peaks at retention time from 12.1 to 16.7 minutes correspond to the odd series from C23 to C29.
|
|
For the quantitative determination of both natural n-alkanes and UCM, the sum of the respective areas, except that of the IS, was considered. On each case those areas were compared
with the area of the IS.
2.4. Method validationTOP
Mineral oil type B (ref. 78473, Fluka, Buchs, Switzerland) was constituted by paraffins from C19 to C41 with maximum at C27.
For de calculation of the response factor, 1 mL portions of this mineral oil in n-hexane were prepared at concentrations of
20, 51, 101, and 507 mg·L–1, by dilution of a 1014 mg·L–1 solution. Refined sunflower oil samples containing 491, 105, 51, 19, and 11 mg·kg–1 mineral oil were prepared by mixing mineral oil type B and a “blank” refined sunflower oil containing less than 5 mg·kg–1 hydrocarbons (method’s LOD).
The IS usually applied for the quantification of the SH was a solution of n-eicosane (C20) in n-hexane at concentration of 0.05 mg·mL–1. This is the smallest natural n-alkane in the majority of vegetable oils. However, in the case of vegetable oils containing significant amounts of natural n-eicosane other n-alkane must be chosen; that is the case of corn oil, where the use of n-nonadecane (C19) is recommended.
The spiked mineral oil solutions were evaporated in a rotary evaporator at room temperature under negative pressure. The residues
were re-dissolved in n-heptane and analysed by GC. Other spiked portions (1 mL) were fractioned through silver-silica gel
column and each time the first 60-mL fraction were analysed by GC.
To evaluate the effect of oil matrix, refined sunflower oil samples containing 491, 105, 51 and 19 mg·kg–1 mineral oil were prepared by blending mineral oil type B and a “blank” refined sunflower oil that yielded an analytical result
below the method’s LOD.
Repeatability at 50 mg·kg–1 level was calculated analysing the oil sample containing 51 mg·kg–1 six times consecutively.
The LOD was determined using the “blank” refined sunflower oil fortified with 10 mg·kg–1 mineral oil.
3. RESULTS AND DISCUSSIONTOP
3.1. Method developmentTOP
3.1.1. Separation of the hydrocarbon fractionsTOP
As pointed out before, to study the behaviour of the different types of hydrocarbons during the separation on silver-silica
gel column, refined olive pomace oil spiked with 3,5-cholestadiene was chosen since it contains significant amounts of squalene
and its derivatives, and steroidal hydrocarbons. The oil (1 g) was fractioned in the silver-silica gel column collecting three
separated fractions of 40, 40, and 20 mL n-hexane, respectively. Subsequently, 120 mL of the n-hexane:diethyl ether (98:2)
mixture were added, collecting three independent 40 mL fractions. Each fraction was analysed by GC. The first 40-mL fraction
contained natural SH and UCM (Figure 1). Negligible amounts of compounds were found in the second 40-mL fraction, whereas a small peak of stigmastadiene appears
in the third 20-mL fraction. The subsequent elution with 40 mL n-hexane:diethyl ether (98:2) extracted cholestadiene and stigmastadiene
and with an additional 40 mL-portion of the same solvent, a complex mixture of hydrocarbons and (aliphatic and terpenic) waxes
was obtained. Finally, the GC chromatogram of a new 40-mL fraction showed alkyl esters of fatty acids and waxes together with
some other humps. Squalene remained in the column. These facts indicate that a very good separation between SH and other compounds
is achieved using silver-silica gel. Elution with 60 mL n-hexane (first fraction) is more than enough to assure a complete
isolation of this fraction without interferences of other compounds, and there is no need to collect further eluates.
3.1.2. Gas chromatography analysis and quantitationTOP
The analysis of the SH fraction coming from a contaminated vegetable oil yields a gas chromatogram showing a broad chromatographic
hump of about 15 min width due to an UCM of branched hydrocarbons typical of mineral oils. On this hump, a series of sharp
peaks corresponding to n-alkanes naturally present in vegetable oils appears. For the quantitative determination of the UCM, a straight baseline was
traced using “manual integration”, from the beginning of the hump (just after the C20 IS has eluted) until the point where
the trace returns to the baseline, resulting in the area of the hump and all the peaks on it (Figure 1A). Next, new valley-valley integrations were performed under each sharp peak appearing on the hump; the areas of all peaks
corresponding to the n-alkane series were added (Figure 1B). The difference between the former and the latter integration results was the area of the UCM. For the determination of
the natural n-alkanes, the sum of the areas of corresponding sharp peaks, except that of the IS, was considered. For quantification, the
areas of the UCM and those of the natural n-alkanes were compared with the area of the IS.
3.2. Method validationTOP
One mL Portions of spiked mineral oil type B solutions were analysed by GC. Other spiked portions (1 mL) were fractioned thorough
silver-silica gel column and analysed also by GC. For concentrations lower than 500 mg·kg–1 response factors with respect to the C20 IS were about 1.02, and recoveries vs. direct analysis, and vs. standard solutions
were higher than 95 and 93%, respectively (Table 1), indicating minimum losses.
Table 1. Responses of direct analysis and recoveries of analysis through silver-silica gel column of mineral oil type B standard
solutions in n-hexane
| Standard solution of mineral oil (mg·L–1)
|
Direct analysis by GC (mg·L–1)
|
Response (%) |
GC analysis after passing through column (mg·L–1)
|
Recovery vs. direct analysis (%) |
Recovery vs. standard solution (%) |
| 1014 |
983 |
96.9 |
909 |
92.5 |
89.6 |
| 507 |
493 |
97.2 |
472 |
95.7 |
93.1 |
| 101 |
98 |
97.0 |
95 |
96.9 |
94.1 |
| 51 |
50 |
98.0 |
49 |
98.0 |
96.1 |
| 20 |
21 |
105.0 |
20 |
95.2 |
100.0 |
To evaluate the effect of oil matrix, “blank” refined sunflower oil samples were mixed at certain concentrations with mineral
oil type B. The analysis of these oils using the proposed method produced recoveries of 93.3, 96.2, 102.1, and 110.5% with
respect to the added mineral oil, and 92.7, 93.5, 96.0, and 95.5% taking into account the content of the “blank oil” (below
5 mg·kg–1). These results indicate that the calibration curve obtained from n-hexane solutions can be used for quantification purposes.
Repeatability at 50 mg·kg–1 level was calculated analysing the oil sample containing 51 mg·kg–1. The standard deviation (SD) was ±1.9 mg·kg–1 and the variation coefficient 4.0%.
A LOD of 5 mg·kg–1 was estimated using the “blank” refined sunflower oil fortified with 10 mg·kg–1 mineral oil. The LOQ was approximately 15 mg·kg–1, three times the LOD, although it depends on the width of hump. This LOQ is sufficient to quantify around the 50 mg·kg–1 limit established by the EU.
The aim of the present work was to develop a reliable and robust analytical method for the detection of mineral and fuel oil
in edible fats and oils, which let compare results to those obtained by other methods. At this point, we participated in the
proficiency test organized by The JRC of the European Commission on the determination of mineral oil in sunflower oils (Joint Research Centre, 2009) where each of the 55 laboratories used its own method. The results obtained using the method proposed in this paper are
shown in Table 2. It can be seen that they are comparable with those obtained by other procedures.
Table 2. Comparison of results of mineral oil determination obtained in the proficiency test organized by the Joint Research
Centre of the EU
|
Results using the proposed method (mg·kg–1)
|
Mean value after removal of outliers (mg·kg–1)
|
Gravimetrically established value (mg·kg–1)
|
| Crude sunflower oil |
381 |
358 |
– |
| Refined sunflower oil |
123 |
113 |
– |
| Refined sunflower oil spiked with mineral oil |
103 |
120 |
114 |
3.3. Collaborative trialTOP
A collaborative trial was carried out with trained Spanish laboratories according to the standard ISO 5725. Samples of “blank”
refined sunflower oil, “blank” samples spiked with 51 mg·kg–1 mineral oil type B, and highly contaminated crude sunflower oil samples were run by nine laboratories following the method
described under Section 2.5, emphasizing the integration requirements. The results are shown in Table 3.
Table 3. Precision data for the determination of various types of saturated hydrocarbons (SH) in refined, spiked and contaminated
crude sunflower oils
|
“Blank” refined sunflower oil |
“Blank” refined sunflower oil spiked with 51 mg·kg–1 mineral oil type B
|
Contaminated crude sunflower oil |
| Total SH |
n-alkanes
|
UCM |
Total SH |
n-alkanes
|
UCM |
Total SH |
n-alkanes
|
UCM |
| Laboratories retained after eliminating outliers |
8 |
8 |
8 |
9 |
9 |
9 |
9 |
9 |
9 |
| Number of test results on sample |
16 |
16 |
16 |
18 |
18 |
18 |
18 |
18 |
18 |
| Mean, mg·kg–1 |
159.6 |
157.9 |
1.6 |
214.2 |
160.3 |
53.9 |
508.1 |
126.1 |
382.0 |
| Repeatability standard deviation (Sr) |
3.4 |
2.8 |
1.3 |
4.2 |
2.6 |
2.7 |
11.9 |
5.0 |
10.1 |
| Repeatability relative standard deviation (RSDr),%
|
2.1 |
1.7 |
81.0 |
1.9 |
1.6 |
5.0 |
2.3 |
4.0 |
2.7 |
| Repeatability limit (r)
|
9.5 |
7.7 |
3.7 |
11.7 |
7.4 |
7.5 |
33.3 |
14.0 |
28.4 |
| Reproducibility standard deviation (SR)
|
7.3 |
6.6 |
2.0 |
14.3 |
8.2 |
8.1 |
30.6 |
6.4 |
30.7 |
| Reproducibility relative standard deviation (RSDR),%
|
4.6 |
4.2 |
122.3 |
6.7 |
5.1 |
14.9 |
6.0 |
5.0 |
8.1 |
| Reproducibility limit (R)
|
20.3 |
18.5 |
5.6 |
39.9 |
22.8 |
22.5 |
85.6 |
17.8 |
86.1 |
| Horrat Index (HoR)
|
0.22 |
0.20 |
2.91 |
0.33 |
0.24 |
0.60 |
0.34 |
0.23 |
0.44 |
A lower precision for determinations of total and UCM hydrocarbons in samples containing a hump of compounds can be observed,
which suggests that the layout of the base line between the beginning and the end of the hump is the main source of error.
This trouble is common to all methods for determining mineral oil using GC analysis.
3.4. Final remarks: GC temperature programmeTOP
In order to improve the precision of the method an increase in the ramp rate of the GC oven temperature was proposed (Moret et al., 2011) since the width of the hydrocarbon hump was reduced; however, special care must be taken to avoid overlapping of this hump
with the possible change of the baseline when the final temperature is reached. The baseline change with 20 °C·min–1 ramp occurs at shorter retention time than with 12 °C·min–1 (Figure 2) producing the alteration of the hump profile appearing in olive-pomace oil analysis (comparing Figures 1 and 3).
|
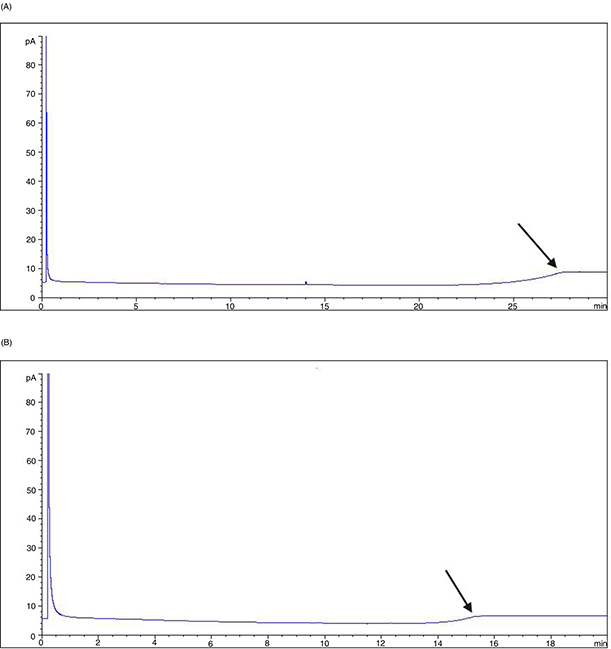 Figure 2. GC baseline profiles of n-heptane obtained at different oven temperature rates: A) 12 °C·min–1; B) 20 °C·min–1. The arrow indicates the baseline change. Figure 2. GC baseline profiles of n-heptane obtained at different oven temperature rates: A) 12 °C·min–1; B) 20 °C·min–1. The arrow indicates the baseline change.
|
|
|
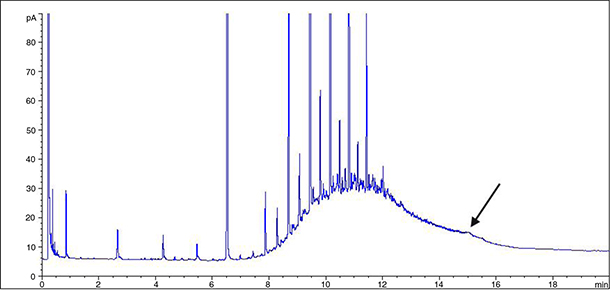 Figure 3. GC profile of the first 40 mL-n-hexane fraction containing SH from refined olive-pomace oil at oven temperature ramp of 20 °C·min–1. The arrow indicates the baseline change when using this temperature ramp. Figure 3. GC profile of the first 40 mL-n-hexane fraction containing SH from refined olive-pomace oil at oven temperature ramp of 20 °C·min–1. The arrow indicates the baseline change when using this temperature ramp.
|
|
4. CONCLUSIONSTOP
The method is reliable for mineral oil detection in the range 10–500 mg·kg–1. At level of the limit established by the EU for mineral oil in sunflower oil (50 mg·kg–1), the repeatability and reproducibility values, and the Horrat index were acceptable (Table 3), and the mean value (53.9 mg·kg–1) was close to the spiking value (51 mg·kg–1). A draft ISO Standard regarding this method is been evaluated for the determination of aliphatic hydrocarbons in vegetable
oils and fats ISO (ISO 2013).
ACKNOWLEDGEMENTSTOP
The Spanish Ministry of Science and Innovation funded this work (ref. AGL2009-07618). The authors would like to thank Ms.
Rosario González Cordones for her assistance in the laboratory.
REFERENCESTOP
| ○ |
AOCS Official Methods of Analysis. Method Ca 6c-65. Hydrocarbons (Mineral oil); Ca 6c-65; AOCS: Champaign, Illinois, USA,
1997, pp 1–2.
|
| ○ |
Bastic M, Bastic L, Jovanovic JA, Spiteller G. 1978. Hydrocarbons and other weakly polar unsaponifiables in some vegetable
oils. J. Am. Oil Chem. Soc. 55, 886–891. http://dx.doi.org/10.1007/BF02671413.
|
| ○ |
Biedermann M, Grob K. 2009a. How “white” was the mineral oil in the contaminated Ukrainian sunflower oils? Eur. J. Lipid Sci. Technol. 111, 313–319 http://dx.doi.org/10.1002/ejlt.200900007.
|
| ○ |
Biedermann M, Fiselier K, Grob K. 2009b. Aromatic hydrocarbons of mineral oil origin in foods: Method for determining the
total concentration and first results. J. Agric. Food Chem. 57, 8711–8721. http://dx.doi.org/10.1021/jf901375e.
|
| ○ |
Biedermann M, Grob K. 2012. On-line coupled high performance liquid chromatography-gas chromatography for the analysis of
contamination by mineral oil. Part 1: Method of analysis. J. Chromatogr. A, 1255, 56–75. http://dx.doi.org/10.1016/j.chroma.2012.05.095.
|
| ○ |
Cert A, Moreda W. 1998. New method of stationary phase preparation for silver ion column chromatography: Application to the
isolation of steroidal hydrocarbons in vegetable oils. J. Chromatogr. A, 823, 291–297. http://dx.doi.org/10.1016/S0021-9673(98)00183-6.
|
| ○ |
EC, 2009. Commission Regulation (EC) No 1151/2009 of 27 November 2009 imposing special conditions governing the import of
sunflower oil originating in or consigned from Ukraine due to contamination risks by mineral oil and repealing Decision 2008/433/EC;
L 313. pp 36–40.
|
| ○ |
Fiorini D, Fiselier K, Biedermann M, Ballini R, Coni E, Grob K. 2008. Contamination of grape seed oil with mineral oil paraffins.
J. Agric. Food Chem. 56, 11245–11250. http://dx.doi.org/10.1021/jf802244r.
|
| ○ |
Fiorini D, Paciaroni A, Gigli F, Ballini R. 2010. A versatile splitless injection GC-FID method for the determination of mineral
oil paraffins in vegetable oils and dried fruit. Food Control, 21, 1155–1160. http://dx.doi.org/10.1016/j.foodcont.2010.01.011.
|
| ○ |
Fiselier K, Fiorini D, Grob K. 2009a. Activated aluminium oxide selectively retaining long chain n-alkanes: Part II. Integration into an on-line high performance liquid chromatography-liquid chromatography-gas chromatography-flame
ionization detection method to remove plant paraffins for the determination of mineral paraffins in foods and environmental
samples. Anal. Chim. Acta. 634, 102–109. http://dx.doi.org/10.1016/j.aca.2008.12.011.
|
| ○ |
Fiselier K, Fiorini D, Grob K. 2009b. Activated aluminium oxide selectively retaining long chain n-alkanes. Part I, description of the retention properties. Anal. Chim. Acta. 634, 96–101. http://dx.doi.org/10.1016/j.aca.2008.12.007.
|
| ○ |
Fiselier K, Grob K. 2009. Determination of mineral oil paraffins in foods by on-line HPLC-GC-FID: lowered detection limit;
contamination of sunflower seeds and oils. Eur. Food Res. Technol. 229, 679–688. http://dx.doi.org/10.1007/s00217-009-1099-8.
|
| ○ |
ISO, 2013. International standard methods for animal and vegetable fats and oils. Method 17780. Determination of Aliphatic
Hydrocarbons; 17780.
|
| ○ |
IUPAC, 1987. Standard Method 2.611 in Standard Methods for the Analysis of Oils, Fats and Derivatives. Determination of mineral
oils in vegetable and animal fats and oils; 2611; Blackwell: Oxford.
|
| ○ |
Joint Research Centre. 2009. Final report on proficiency test on the determination of mineral oil in sunflower oil. |
| ○ |
Lanzón A, Albi T, Cert A, Gracián J. 1994. The hydrocarbon fraction of virgin olive oil and changes resulting from refining.
J. Am. Oil Chem. Soc. 71, 285–291. http://dx.doi.org/10.1007/BF02638054.
|
| ○ |
Moret S, Barp L, Grob K, Conte LS. 2011. Optimised off-line SPE-GC-FID method for the determination of mineral oil saturated
hydrocarbons (MOSH) in vegetable oils. Food Chem. 129, 1898–1903. http://dx.doi.org/10.1016/j.foodchem.2011.05.140.
|
| ○ |
Moret S, Populin T, Conte LS, Grob K, Neukom HP. 2003. Occurrence of C15-C45 mineral paraffins in olives and olive oils. Food Addit. Contam. 20, 417–426. http://dx.doi.org/10.1080/0265203031000098687.
|
| ○ |
Neukom HP, Grob K, Biedermann M, Noti A. 2002. Food contamination by C20-C50 mineral paraffins from the atmosphere. Atmos. Environ. 36, 4839–4847. http://dx.doi.org/10.1016/S1352-2310(02)00358-8.
|
| ○ |
Populin T, Biedermann M, Grob K, Moret S, Conte L. 2004. Relative hopane content confirming the mineral origin of hydrocarbons
contaminating foods and human milk. Food Addit. Contam. 21, 893–904. http://dx.doi.org/10.1080/02652030400001164.
|
| ○ |
Tan YA, Kuntom A. 1993. Gas chromatographic determination of hydrocarbons in crude palm kernel oil. J. Assoc. Off. Anal. Chem. 76, 371–376.
|
| ○ |
Tranchida PQ, Zoccali M, Purcaro G, Moret S, Conte L, Beccaria M, Dugo P, Mondello L. 2011. A rapid multidimensional liquid-gas
chromatography method for the analysis of mineral oil saturated hydrocarbons in vegetable oils. J. Chromatogr. A, 1218, 7476–7480. http://dx.doi.org/10.1016/j.chroma.2011.06.089.
|
| ○ |
Wagner C, Neukom HP, Grob K, Moret S, Populin T, Conte LS. 2001. Mineral paraffins in vegetable oils and refinery by-products
for animal feeds. Mitt. Lebensmittelunters. Hyg. 92, 499–514.
|
| ○ |
Wagner C, Neukom HP, Galetti V, Grob K. 2001. Determination of mineral paraffins in feeds and foodstuffs by bromination and
preseparation on aluminium oxide: method and results of a ring test. Mitt. Lebensmittelunters. Hyg. 92, 231–249.
|