n-3 LCPUFA in the reversal of hepatic steatosis: the role of ACOX and CAT-1
G.S. Tapia, D. González-Mañán, A. D’Espessailles and C.G. Dossi*
Molecular and Clinical Pharmacology Program, Institute of Biomedical Sciences, Faculty of Medicine, University of Chile, Santiago,
Chile
*Corresponding author: camila.g.dossi@ug.uchile.cl
| |
SUMMARY
The aim of this study was to investigate the roles of the Acyl co-enzyme A oxidase (ACOX), carnitine acyl transferase I (CAT-1)
and activating protein 1 (AP-1) in the reversal of hepatic steatosis with dietary change and n-3 long chain polyunsaturated fatty acid (n-3 LCPUFA) supplementation. Male C57BL/6J mice were given either a control diet (CD) or a high fat diet (HFD) for 12 weeks,
and then continued with the CD or CD plus n-3 LCPUFA for eight weeks. After this period, body and adipose visceral tissue weight were analyzed and liver samples were taken
to measure ACOX, CAT-1 and c-jun levels. The dietary change from HFD to a norm caloric diet plus n-3 LCPUFA supplementation significantly reduced liver steatosis and adipose tissue: body weight ratio, along with an increase
in the hepatic ACOX and CAT-1 levels and normalization of AP-1 expression that could favor the fatty acid beta-oxidation over
lipogenesis and regulate inflammation. These results provide new data on the enzymatic metabolism underlying dietary change
to a norm caloric diet plus n-3 LCPUFA supplementation.
|
| |
RESUMEN
n-3 AGPICL en la reversión de la esteatosis hepática: el papel de la ACOX y CAT. El objetivo de este estudio fue investigar el rol de las enzimas Acil coenzima A oxidasa (ACOX) y Acil carnitina transferasa
1 (CAT-1), además del factor de transcripción, Proteína activadora 1 (AP-1) en la reversión de la esteatosis hepática mediante
cambio de dieta más suplementación con Ácidos grasos poliinsaturados de cadena larga omega tres (AGPICL n-3). Ratones macho
de la cepa C57BL/6J fueron alimentados con dieta control (DC) o alta en grasas (DAG) durante 12 semanas, luego continuaron
con DC con o sin suplementación de AGPICL n-3 durante 8 semanas. Después de este período, se analizó el peso corporal y del
tejido adiposo visceral; en las muestras hepáticas se evaluaron los niveles de ACOX, CAT-1 y AP-1. El cambio a dieta control
más suplementación con AGPICL n-3 reduce significativamente la esteatosis hepática y la relación tejido adiposo/peso corporal,
acompañado de un incremento en los niveles hepáticos de ACOX y CAT-1 y normalización de la expresión de AP-1; regulando la
inflamación y favoreciendo la beta-oxidación sobre la lipogénesis. Estos resultados proveen nuevos datos sobre el metabolismo
enzimático cuando se realiza cambio a dieta control más suplementación con AGPICL n-3.
|
1. INTRODUCTIONTOP
The non-alcoholic fatty liver disease (NAFLD) is considered to be the hepatic expression of the metabolic syndrome, defined
as the clustering of risk factors for cardiovascular disease and type II diabetes, which include hyperglycemia, insulin resistance,
hypertriglyceridemia and obesity (Bellentani et al., 2009; Malaguarnera et al., 2009). It is the most prominent cause of chronic liver disease and it is characterized by a intra-hepatic triacylglycerol (TG)
content higher than 5% of the liver weight (steatosis) in the absence of significant alcohol consumption (10–20g/day in women;
20–30 g/day in men) (Musso et al., 2009), which can progress to inflammation (steatohepatitis), fibrosis, and cirrhosis (Valenzuela et al., 2011). The pathogenic mechanisms involved in the development of hepatic steatosis are not completely understood, but it is known
that it results from an imbalance between lipid availability, either from enhanced uptake and/or de novo lipogenesis, and
lipid disposal, either from decreased mitochondrial and peroxisomal fatty acid oxidation and/or reduced lipid output by the
liver (Musso et al., 2009). The establishment of liver steatosis leads to the production of free radicals with a lipid peroxidation response, pro-inflammatory
cytokine release (Aronis et al., 2005), and n-3 long-chain polyunsaturated fatty acid (n-3 LCPUFA) depletion, with enhancement in the n-6/n-3 LCPUFA ratio favoring a pro-inflammatory state. This n-3 LCPUFA depletion induces changes in the DNA binding activity of the peroxisome proliferator-activated receptor Alfa (PPAR-α) (Valenzuela et al., 2011), with a decrease in PPAR-α phosphorylation thus eliciting low transcription levels of the target genes for carnitine
acyl transferase 1 (CAT-1) and acyl coenzyme A oxidase (ACOX), favoring lipogenesis over fatty acid oxidation (Araya et al., 2004; Araya et al., 2010). CAT-1 is an enzyme that regulates the process of beta oxidation in mitochondria, while ACOX is responsible for fatty acid
dehydrogenation to promote the beta oxidation process in the peroxisomes (Barlett et al., 2004; Poirier et al., 2006).
Secondly, it has been shown that in NAFLD the DNA binding of c-jun (AP-1 subunit) is increased, enhancing the production of
pro-inflammatory cytokines, which promote an inflammation state and facilitate the development and progression of the disease
(Dorn et al., 2014).
The transcriptional factor AP-1 is a protein complex of two subunits from families Jun (c-jun, junB and junD) and Fos (c-Fos,
FosB, among others) forming homo-dimers or hetero-dimers between them (Halazonetti et al., 1988). AP-1 is capable of promoting cytokine expressions such as TNF-α (tumor necrosis Alfa factor), IL (interleukin)-1
and 2: which are involved in the recruitment and activation of Kupffer cells and the progression of NAFLD (Baffy, 2009).
Because of the increasing prevalence of NAFLD, numerous studies have focused on therapeutic strategies for this liver disease
which have included weight loss, physical activity, and therapies based on insulin sensitizers, lipid-lowering agents, antioxidant
drugs and n-3 LCPUFAs (Nobili et al., 2012).
Previous studies by our group have demonstrated that n-3 LCPUFA supplementation prevents the pro-steatotic and pro-inflammatory effects of a high-fat diet (HFD) at the hepatic level
(Valenzuela et al., 2012). Furthermore, the dietary change from HFD to a norm caloric diet with n-3 LCPUFA supplementation significantly reduced insulin resistance and liver steatosis when compared to switching HFD to a norm
caloric diet alone (Dossi et al., 2014).
The aim of this study was to investigate the role of ACOX, CAT-1 and AP-1 in the reversal of hepatic steatosis with dietary
change and n-3 LCPUFA supplementation. Parameters related to liver morphological characteristics (lipid vesicles), metabolic
syndrome (visceral adipose tissue), liver total fat content, enzymes involved in lipid metabolism as ACOX and CAT-1 (western
blot and PCR), and the levels of the AP-1 c-jun subunit (immunohistochemistry) were determined.
2. MATERIALS AND METHODSTOP
2.1. Ethics statementTOP
Experimental animal protocols and animal procedures complied with the Guide for the Care and Use of Laboratory Animals (National
Academy of Sciences, NIH Publication 6–23, revised 1985) and were approved by the Bioethics Committee for Research in Animals,
Faculty of Medicine, University of Chile (CBA 0386 FMUCH).
2.2. Animal preparation and supplementation with n-3 LCPUFA (EPA plus DHA)TOP
Weaning male C57BL/6J mice weighing 12 to 14 g were obtained from the Animal Facility at the Faculty of Medicine, University
of Chile, Santiago, Chile. Room temperature was kept constant at 21 °C and light was maintained on a 12:12-h light-dark cycle.
At 20 days of age, mice were randomly divided into two diet groups: i) the control diet containing (wt/wt) 10% fat, 20% protein,
and 70% carbohydrate or ii) a high-fat diet (HFD) containing (wt/wt) 60% fat, 20% protein, and 20% carbohydrate (D12492, Research
Diets, NJ, USA) from days 1 to 84 (12 weeks). 60% of the fat in the HFD was provided by the addition of lard (saturated fatty
acids and cholesterol) in significantly higher amounts compared to the control diet. After 12 weeks, the animals given the
control diet were divided into two diet groups (n=9): a) control diet, b) control diet plus n-3 LCPUFA; similarly, the animals subjected to the HFD were divided into two diet groups (n=9): c) change to control diet
and d) change to control diet plus n-3 LCPUFA; all groups continued during 8 weeks to complete 20 weeks of total treatment. The n-3 LCPUFA supplemented groups received fish oil (encapsulated fish oil containing 200 mg·kg–1 [108 mg·kg–1 of EPA and 92 mg·kg–1 of DHA]; Acolest) through oral administration; the control groups were given iso-volumetric amounts of saline. Weekly controls
of body weight and diet intake were carried out throughout the entire period. At the end of the 20th week, the animals were fasted (6–8 h) and then given an anesthesia with tiletamine and zolacepam (Zoletil®, Virbac Laboratories).
The visceral adipose tissue was extracted from the epididymal area, weighed and stored at −20 °C.
2.3. Tissue and blood samplesTOP
Liver samples were frozen in liquid nitrogen and stored at −80 °C, or fixed in phosphate-buffered formalin, embedded in paraffin,
sectioned using a microtome and stained with haematoxylin-eosin. Blood samples were taken from a cardiac puncture, and then
centrifuged, and the serum was stored at −20 °C. Liver slides were stained with haematoxylin-eosin (HE) assessed by optical
microscopy (Olympus CX31, Japan) for morphology analysis in a blind fashion. The presence of both steatosis and inflammation
were graded as absent, mild, moderate or severe (Brunt et al., 1999).
2.4. Western blot analysis of CAT-1 and AP-1TOP
Liver samples (100–500 mg) frozen in liquid nitrogen were homogenized and suspended in a buffer solution pH 7,9 containing
10mM HEPES, 1mM EDTA, 0,6% Nonidet p-40, 150mM NaCl and protease inhibitors (1mM phenylmethylsulfonyl fluoride, 1 μg·mL–1 aprotinin, 1 μg·mL–1 leupeptin, and 1mM orthovanadate). Soluble protein fractions (10 mg) were separated on 12% polyacrylamide gel electrophoresis
using SDS-PAGE, transferred to nitrocellulose membranes, and blocked with TBS containing 5% non-fat dry milk for 1 h at room
temperature. The blots were washed with TBS-containing 0.1% Tween 20 and hybridized with rabbit polyclonal antibodies for
human CAT-1 (Santa Cruz Biotechnology, CA, USA) and anti-(c-Jun) (Fitzgerald Industries International, Acton, MA, U.S.A.).
Mouse monoclonal antibody for rat β-actin (ICN Biomedicals, Inc., Aurora, OH) was used as internal control in all determinations.
After extensive washing, the antigen-antibody complexes were detected using labeled horseradish peroxidasa, and a Super Signal
West Pico Chemiluminescence kit detection system (Pierce, Rockford, IL, USA).
2.5. RT-PCR assay of ACOX mRNA expressionTOP
The expression of ACOX was assessed by RT-PCR. Total RNA was isolated from 15–25 mg of frozen liver using an E.Z.N.A. total
RNA Kit (Omega Biotek, Norcross, Georgia, USA) according to the manufacturer’s instructions. Quantification of total RNA was
performed spectrophotometrically (A260/A280 ratio) and RNA quality was checked by electrophoresis on 1.2% agarose gels, using
a molecular size marker. The resulting DNAse free RNA was reverse-transcribed to cDNA with a Thermo Script reverse transcriptase
(Invitrogen Corp., Carlsbad, CA, USA) according to the manufacturer’s instructions using random hexamer primers (Promega,
Madison, WI, USA). The resulting cDNA was amplified in a PCR reaction using Taq DNA polymerase recombinant (Invitrogen Corp.,
Carlsbad, CA, USA), according to the manufacturer’s instructions. Nucleotide sequences for sense and antisense primers used
in this study were (Fwd: 5`-ACCTTCAGGCCCAAGTGAGT-3` and Rv : 5`-GAGCCCCTGTGATGATGTTC-3`) and the control 18S rRNA as internal
control (Quantum RNA 18S Internal Standards, Ambion Inc., Austin TX, EE.UU). The amplification (TECHNE TC-5000, Bibby Scientific
Ltd., United Kingdom) was initiated after 5 min de-naturation at 94 °C, followed by 34 cycles (94 °C for 30s, 62 °C for 30
s, 72 °C for 30 s) and finalizing with 72 °C for 5 min. The expected outputs for each amplification correspond to 249 pb for
ACOX. All amplification products were stored at 4 °C. PCR products were electro-phoresed on 1.2% agarose gels containing ethidium
bromide, visualized by UV-induced fluorescence, and analyzed for densitometry using UN-SCAN-IT software (Silk Scientific Inc.,
Orem, UT, USA).
2.6. Immunohistochemistry studiesTOP
For AP-1 staining a specific antibody was used against the c-jun subunit followed by deparaffination and rehydration. The
sections were then incubated with EDTA for 45 minutes at 95 °C. Then, blocking was carried out for 30 minutes followed by
incubation with primary antibody anti-(c-jun) (Fitzgerald Industries International, Acton, MA, U.S.A) according to the manufacturer’s
instruction. For nuclear contrast the Hematoxilin of Mayer was used (Modified solution according to Lillie, ScyTek Laboratories,
Utah, USA). An analysis of positive nuclei was performed under light microscope in a blind fashion in 10 adjacent (400×) per
slide. Then a percentage of positive nuclei per slide was calculated.
2.7. Statistical analysesTOP
Statistical analysis was performed with GraphPad PrismTM version 5.0 (GraphPad Software, Inc. San Diego, CA, USA). Values shown represent the mean ± SEM for the number of separate
experiments indicated. One-way ANOVA and the Newman-Keuls test for unpaired data assessed the statistical significance of
differences between mean values as indicated, considering P<0.05 as significant.
3. RESULTSTOP
3.1. Reduction in adipose tissue weight and corporal weight ratio, liver steatosis and morphological alterations induced by
a HFD with change to the control diet with n-3 LCPUFA supplementationTOP
The weights of the animals subjected to the control diet or HFD with and without n-3 LCPUFA supplementation were not significantly
different. However, the visceral adipose tissue/body weight ratio was enhanced (P<0.05) in the mice subjected to HFD+CD compared with CD+CD, CD+ CD/n-3 LCPUFA, and HFD+CD/n-3 LCPUFA (Fig. 1A). In all groups, liver histology was characterized by the absence of architectural distortion, lobular inflammation, necrotic
foci, or fibrosis (Fig. 1B), and those given CD with and without n-3 LCPUFA did not show lipid infiltration [Fig. 1B, (a) and (b)]. However, HFD+CD exhibited macro and micro-vesicular steatosis (30% lipid vesicles infiltration) (Fig. 1B (c)), whereas HFD+CD/n-3 LCPUFA elicited 8% fat infiltration (Fig. 1B (d); Fig. 1C; P<0.05).
|
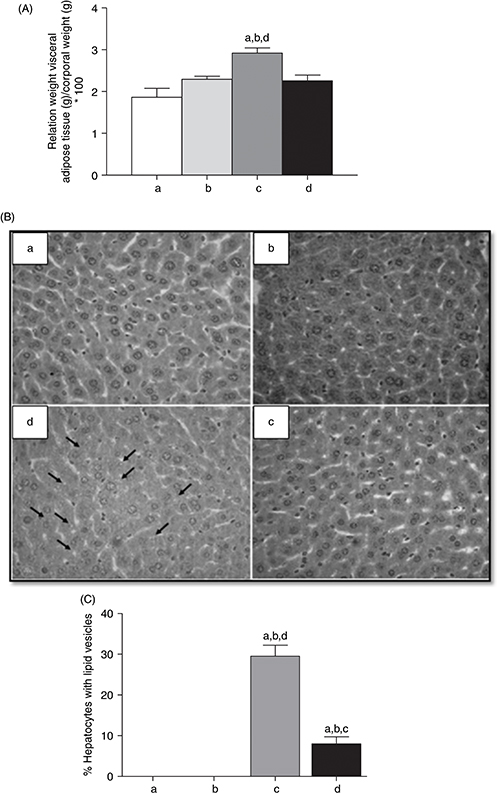 Figure 1. Effect of n-3 long-chain polyunsaturated fatty acid (n-3 LCPUFA) and dietary change on (A) relation between weight of visceral adipose tissue (g) and corporal weight (g), (B) liver
histology (magnification×100) and (C) hepatocyte lipid infiltration in mice. Animals were given either (a) control diet (CD),
(b) CD plus n-3 LCPUFA (CD+CD/n-3 LCPUFA), (c) high fat diet (HFD) followed by CD (HFD+CD), or (d) HFD plus CD supplemented with n-3 LCPUFA (HFD+CD/n-3 LCPUFA). Values are expressed as mean±SEM for 9 animals per experimental group. Letters above the bars indicate statistically
significant differences (P<0.05) compared to the respective animals given the control diet assessed by one-way ANOVA and the Newman-Keuls. Figure 1. Effect of n-3 long-chain polyunsaturated fatty acid (n-3 LCPUFA) and dietary change on (A) relation between weight of visceral adipose tissue (g) and corporal weight (g), (B) liver
histology (magnification×100) and (C) hepatocyte lipid infiltration in mice. Animals were given either (a) control diet (CD),
(b) CD plus n-3 LCPUFA (CD+CD/n-3 LCPUFA), (c) high fat diet (HFD) followed by CD (HFD+CD), or (d) HFD plus CD supplemented with n-3 LCPUFA (HFD+CD/n-3 LCPUFA). Values are expressed as mean±SEM for 9 animals per experimental group. Letters above the bars indicate statistically
significant differences (P<0.05) compared to the respective animals given the control diet assessed by one-way ANOVA and the Newman-Keuls.
|
|
3.2. Change to control diet with n-3 LCPUFA supplementation reduces the liver expression of enzymes involved in fatty oxidationTOP
Measurement of the enzymes involved in fatty liver oxidation CAT-1 and ACOX (Fig. 2) showed that mice subjected to the change from HFD to CD had lower expressions in their mRNA (ACOX, Fig. 2A) and protein levels (CAT-1) (Fig. 2B) with respect to the CD group (P<0.05), whereas the change from HFD to CD/n-3 LCPUFA increased the expression of these enzymes to control values (Fig. 2A y B).
|
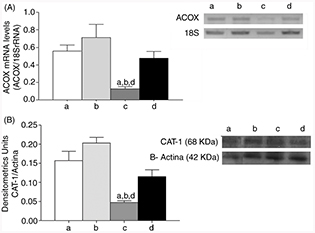 Figure 2. Effect of n-3 long-chain polyunsaturated fatty acid (n-3 LCPUFA) and dietary change on expression of liver enzymes Acyl coenzyme A oxidase (ACOX) mRNA levels (A) and Carnitina acyl
transferase 1 (CAT-1) (Western blot). Animals were given either (a) control diet (CD), (b) CD plus n-3 LCPUFA (CD+CD/n-3 LCPUFA), (c) high fat diet (HFD) followed by CD (HFD+CD), or (d) HFD plus CD supplemented with n-3 LCPUFA (HFD+CD/n-3 LCPUFA). Values are expressed as mean±SEM for 9 animals per experimental group. Letters above the bars indicate statistically
significant differences (P<0.05; one-way ANOVA and the Newman-Keuls, test). Figure 2. Effect of n-3 long-chain polyunsaturated fatty acid (n-3 LCPUFA) and dietary change on expression of liver enzymes Acyl coenzyme A oxidase (ACOX) mRNA levels (A) and Carnitina acyl
transferase 1 (CAT-1) (Western blot). Animals were given either (a) control diet (CD), (b) CD plus n-3 LCPUFA (CD+CD/n-3 LCPUFA), (c) high fat diet (HFD) followed by CD (HFD+CD), or (d) HFD plus CD supplemented with n-3 LCPUFA (HFD+CD/n-3 LCPUFA). Values are expressed as mean±SEM for 9 animals per experimental group. Letters above the bars indicate statistically
significant differences (P<0.05; one-way ANOVA and the Newman-Keuls, test).
|
|
3.3. Change to control diet with n-3 LCPUFA supplementation normalizes the AP-1 levels.TOP
The presence of liver AP-1 in nuclear fractions, assessed by immunohistochemistry, (Fig. 3A), revealed that the mice subjected to CD+CD and CD+CD/n-3 LCPUFA exhibited a percentage of 36.53% and 33.13% of c-jun liver positive nuclei, respectively; whereas animals given HFD+CD showed 63.30% (P<0.05). The HFD+CD/n-3 LCPUFA group shows a diminished c-jun liver positive nuclei (P<0.05) with respect to HFD+CD group, with a 44.30% of c-jun liver positive nuclei (Fig. 3B). However, when analyzed by western blot, there were no significant differences at the c-jun protein level among all the
groups (Fig. 3C).
|
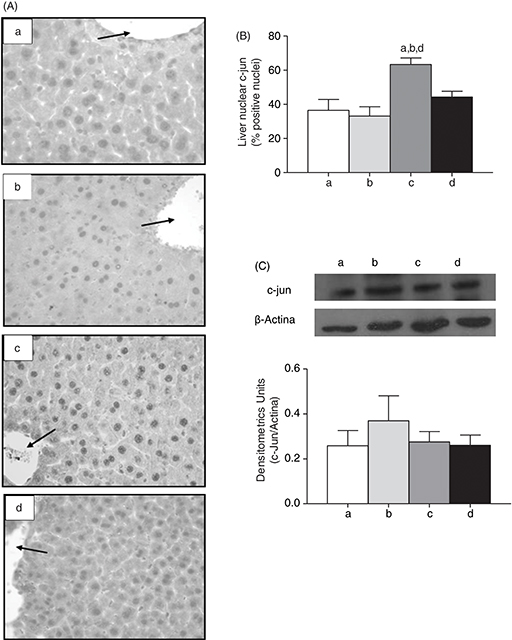 Figure 3. Effect of n-3 long-chain polyunsaturated fatty acid (n-3 LCPUFA) and dietary change on (A) immunohistochemical determination of c-jun nuclear, (B) percentage of positive nuclei for
c-jun, and (C) densitometric units of c-jun (Western blot) in mice. Animals were given either (a) control diet (CD), (b) CD
plus n-3 LCPUFA (CD+CD/n-3 LCPUFA), (c) high fat diet (HFD) followed by CD (HFD+CD), or (d) HFD plus CD supplemented with n-3 LCPUFA (HFD+CD/n-3 LCPUFA). Values are expressed as mean±SEM for 9 animals per experimental group. Letters above the bars indicate statistically
significant differences (P<0.05; one-way ANOVA and the Newman-Keuls test). Figure 3. Effect of n-3 long-chain polyunsaturated fatty acid (n-3 LCPUFA) and dietary change on (A) immunohistochemical determination of c-jun nuclear, (B) percentage of positive nuclei for
c-jun, and (C) densitometric units of c-jun (Western blot) in mice. Animals were given either (a) control diet (CD), (b) CD
plus n-3 LCPUFA (CD+CD/n-3 LCPUFA), (c) high fat diet (HFD) followed by CD (HFD+CD), or (d) HFD plus CD supplemented with n-3 LCPUFA (HFD+CD/n-3 LCPUFA). Values are expressed as mean±SEM for 9 animals per experimental group. Letters above the bars indicate statistically
significant differences (P<0.05; one-way ANOVA and the Newman-Keuls test).
|
|
4. DISCUSSIONTOP
NAFLD is a condition that requires multiple factors for development (Adams et al., 2005), which are induced by excessive carbohydrate or lipid dietary ingestion (Leamy et al., 2013). Once this condition is acquired, it is important to slow its progress to steatohepatitis; later the condition is essentially
irreversible (Dowman et al., 2010).
Mice with the HFD intake developed glucose intolerance and insulin resistance, with higher visceral adiposity and hepatic
steatosis when compared with animals subjected to a norm caloric diet (Dossi et al., 2014). Previously published data show that a norm caloric diet plus n-3 LCPUFA supplementation reverses the HFD-induced pro-steatotic and pro-inflammatory state in mice livers. This reversal was
evidenced by a reduction in insulin resistance, suppression of liver oxidative stress and inflammatory cytokine expression
(IL-1β and TNF-α). These findings were associated with major changes in hepatic nuclear abundance of the lipid metabolism-related
transcription factors PPAR-α and SREBP-1c, with abolishment of the HFD-induced enhancement in SREBP-1c/PPAR-α ratios by combined
HFD and n-3 LCPUFA supplementation. This situation favors catabolism over anabolism and that effect could be mediated by SREBP-1c and
PPAR-α (Dossi et al., 2014). This view is supported by the significant correlation between the reduction observed in the hepatic levels of the PPAR-α-regulate
enzymes ACOX and CAT-1 (Figure 2A y 2B). From a mechanistic point of view, down-regulation of liver enzymes ACOX and CAT-1 may be ascribed to hepatic n-3 LCPUFA depletion (Araya et al., 2004; Araya et al., 2010; Valenzuela et al., 2012) observed in experimental and human obesity. Different PPAR-α agonist factors are able to lower the number of available triglycerides
through different pathways (Araya et al., 2004; Surwit et al., 1988). In this respect, here we observed a significant increase in both mRNA ACOX levels and CAT-1 protein levels in those animals
that underwent the norm caloric diet change and supplementation with n-3 LCPUFA.
The effects of n-3 LCPUFA could be due to several molecular mechanisms. These include a decrease in fatty acids and glycerol mobilization from
peripheral tissue lipolisis to the liver (Valenzuela et al., 2012). In addition, n-3 LCPUFA enhances the antioxidant potential of the liver, acting through direct (ROS scavenging) and/or indirect (nuclear transcription
factor erythroid 2 related factor 2 (Nrf2) activation) mechanisms, a condition proposed to improve insulin sensitivity (De
Leijer et al., 2010; Fernández-Sanchez et al., 2011). Alternatively, the n-3 LCPUFA metabolism generates various potent anti-inflammatory mediators including: i) E-series and D-series of resolvins produced
by the action of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) (Michalik et al., 2006), ii) 5-LOX-dependent protectin D1 production (Videla et al., 2012) and iii) formation of epoxyeicosaquatraenoic acid and epoxydocosapentaenoic acid catalyzed by cytochrome P450 NADPH-dependent
epoxygenases (Houstis et al., 2006). n-3 LCPUFA effects on liver fibrosis and inflammation are being currently addressed by several clinical trials (De Roos et al., 2009).
Previous studies showed that these metabolic alterations decrease when changed to a norm caloric diet (Dossi et al., 2014; Surwit et al., 1988; De Meijer et al., 2010), whereas n-3 LCPUFA supplementation prevented an HFD-induced increase in liver lipid content (Nobili et al., 2012; Valenzuela et al., 2012). The increase in visceral adipose tissue content induced by the HFD produces an increment in the pro-inflammatory cytokine (adipokines) secretion such as IL-6, IL-1β and TNF-α (Fernández-Sánchez et al., 2011). In this respect, the significant increase in the visceral adipose tissue/corporal body weight ratio and hepatic lipid infiltration in the the HFD+CD group was normalized in the HFD+CD/n-3 LCPUFA group, meaning that n-3 LCPUFAS are involved in lipid content changes and thus could be also involved in the cytokine expression decrease. The decrease in the cytokine expression could be explained by the reduction in the nuclear c-jun content. Previous studies support the participation of c-jun (as a subunit of AP-1) in developing NAFLD, supporting the results found in this study (Dorn et al., 2014). The increase in AP-1 expression could be in part responsible for the pro-inflammatory cytokine (IL-1β and TNF-α) expression observed in HFD (Dossi et al., 2014). It has been observed that increasing c-jun, and therefore AP-1, leads to an increased transcription of the c-jun, in an
auto-regulatory cycle, indicating that the c-jun increase in NAFLD is a consistent indicator of the AP-1 dimmer increase,
and it could participate in increasing the transcription of pro-inflammatory cytokines, which are elevated in the development
of this pathology (Hasenfuss et al., 2014). In another aspect, the action of AP-1 factor has been linked with the sensor lipid PPAR, postulating an inhibitory role
to the AP-1 factor, acting in the PPAR-DNA binding (Ye et al., 2002); thus, performing a protective role. In mice with HFD-induced hepatic steatosis an upregulation of PPAR-γ levels was evident
(Shapiro et al., 2011) and is regulated differently depending on the AP-1 dimer formed: c-jun joined Fra-1 and Fra-2 (members of the Fos family)
inhibit the expression of PPAR-γ resulting in a liver steatotic reduction, but the dimmer formed by c-jun and c-Fos has the
opposite effect (Fernández-Sanchez et al., 2011).
To increase the reliability of the results obtained in this work, we analyzed the subunit c-jun levels using two techniques
based on the antigen-antibody union: immunohistochemistry and western blotting; both techniques aim to determinate the relative
levels of the protein. The immunohistochemical analysis allowed for determination of the AP-1 levels through the measurement
of the subunit c-jun (Halazonetis et al., 1988; Hong et al., 2003). The analysis showed that dietary change associated with n-3 LCPUFA supplementation is effective in reducing AP-1 levels, although it does not show significant differences in the measurement
by western blot. Regarding the differences obtained with both techniques, it is important to mention that tissue processing
is a relevant factor in the discrepancy found in the results of each technique. Immunohistochemistry is set to maintain the
morphology of the living body, which involves maintaining the three dimensional structure of protein; while in the western
blotting, the tissue is homogenized and then treated with de-naturating agents such as SDS (sodium dodecyl sulfate) and β-mercaptoethanol,
causing a loss of the three-dimensionality of proteins, which could directly affect the antigen-antibody binding agents. The
loss of the tertiary structure of c-jun may be the reason for the differences in the results between the two techniques as
antibody binding can occur in the different structures that make up a protein (primary, secondary, tertiary or quaternary).
Considering these results together, we conclude that n-3 LCPUFA supplementation associated with diet change reverses the HFD-induced hepatic steatosis, along with a recovery of ACOX
and CAT-1 levels and normalization of the AP-1 nuclear subunit c-jun levels.
FUNDING SOURCESTOP
The studies carried out in the Laboratory were funded by Grant 1110043 or 1140547 (to G.T) from Fondo Nacional de Desarrollo
Científico y Tecnológico- FONDECYT (Chile).
REFERENCESTOP
| ○ |
Adams LA, Angulo P and Lindor KD. 2005. Nonalcoholic fatty liver disease. Can. Med. Assoc. J. 172, 899–905. http://dx.doi.org/10.1503/cmaj.045232.
|
| ○ |
Angel P, Hattori K, Smeal T and Karin M. 1988. The jun proto-oncogene is positively autoregulated by its product, jun/AP-1.
Cell. 55, 875–885. http://dx.doi.org/10.1016/0092-8674(88)90143-2.
|
| ○ |
Araya J, Rodrigo R, Pettinelli P, Araya V, Poniachik J and Videla L. 2010. Decreased liver fatty acid Δ-6 and Δ-5 desaturase
activity in obese patients. Obesity. 18, 1460–1463. http://dx.doi.org/10.1038/oby.2009.379.
|
| ○ |
Araya J, Rodrigo R, Videla L, Thielemann L, Orellana M, Pettinelli P and Poniachik J. 2004. Increase in long-chain polyunsaturated
fatty acid n-6/n-3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin. Sci. 106, 635–643. http://dx.doi.org/10.1042/cs20030326.
|
| ○ |
Aronis A, Madar Z and Tirosh O. 2005. Mechanism underlying oxidative stress-mediated lipotoxicity: Exposure of J774.2 to macrophages
triacylglycerols facilitates mitochondrial reactive oxygen species production and cellular necrosis. Free Radic. Biol. Med. 38, 1221–1230. http://dx.doi.org/10.1016/j.freeradbiomed.2005.01.015.
|
| ○ |
Baffy G. 2009. Kupffer cells in non-alcoholic fatty liver disease: The emerging view. J. Hepatol. 51, 212–223. http://dx.doi.org/10.1016/j.jhep.2009.03.008.
|
| ○ |
Bartlett K and Eaton S. 2004. Mitochondrial β-oxidation. Eur. J. Biochem. 271, 462–469. http://dx.doi.org/10.1046/j.1432-1033.2003.03947.x.
|
| ○ |
Bellentani S, Scaglioni F, Marino M and Bedogni G. 2010. Epidemiology of non-alcoholic fatty liver disease (NAFLD). Dig Dis. 28, 155–161. http://dx.doi.org/10.1159/000282080.
|
| ○ |
Brunt EM, Janney CG, Di Biscegle AM, Neuschwander-Tetri BA and Bacon BR. 1999. Nonalcoholic steatohepatitis: a proposal for
grading and staging the histological lesions. Am. J. Gastroenterol. 94, 2467–2474. http://dx.doi.org/10.1111/j.1572-0241.1999.01377.x.
|
| ○ |
Delerive P, Fruchart JC and Staels B. 2001. Peroxisome proliferator-activated receptors ininflamation control. J. Endocrinology. 169, 453–459. http://dx.doi.org/10.1677/joe.0.1690453.
|
| ○ |
De Roos B, Mavrommatis Y and Brouwer I. 2009. Long-chain n-3 polyunsaturated fatty acids: new insights into mechanisms relating
to inflammation and coronary heart disease. Br. J. Pharmacol. 158, 413–428. http://dx.doi.org/10.1111/j.1476-5381.2009.00189.x.
|
| ○ |
De Meijer V, Le H, Meisel J, Sharif M, Pan A, Nosé V and Puder M. 2010. Dietary fat intake promotes the development of hepatic
steatosis independently from excess caloric consumption in a murine model. Metabolism. 59, 1092–105. http://dx.doi.org/10.1016/j.metabol.2009.11.006.
|
| ○ |
Dorn C, Engelmann J, Saugspier M, Koch A, Hartmann A, Müller M, Spang R, Bossenhorff A and Hellerbrand C. 2014. Increased
expression of c-Jun in nonalcoholic fatty liver disease. Lab Invest. 94, 394–408. http://dx.doi.org/10.1038/labinvest.2014.3.
|
| ○ |
Dossi CG, Tapia GS, Espinosa A, Videla LA and D’Espessailles A. 2014. Reversal of High-fat diet-induced hepatic steatosis
by n-3 LCPUFA: role of PPAR-α and SREBP-1c. J. Nutr. Biochem. 25, 977–984. http://dx.doi.org/10.1016/j.jnutbio.2014.04.011.
|
| ○ |
Dowman JK, Tomlinson JW and Newsome PN. 2010. Pathogenesis of non-alcoholic fatty liver disease. Int. J. Med. 103, 71–83. http://dx.doi.org/10.1093/qjmed/hcp158.
|
| ○ |
Fernandez-Sanchez A, Madrigal-Santillan E, Bautista M, Esquivel-Soto J, Morales-Gonzalez A, and Esquivel-Chirino C. 2011.
Inflammation, Oxidative Stress, and Obesity. Int J Mol Sci. 12, 3117–32. http://dx.doi.org/10.3390/ijms12053117.
|
| ○ |
Hasenfuss SC, Bakiri L, Thomsen M, Williams E, Auwerx J and Wagner E. 2014. Regulation of Steatohepatitis and PPARγ Signaling
by Distinct AP-1 Dimers. Cell Metabolism. 19, 84–95. http://dx.doi.org/10.1016/j.cmet.2013.11.018.
|
| ○ |
Hong S, Gronert K, Devchand PR, Moussignac RL and Serhan CN. 2003. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic
acid in murine brain, human blood, and glial cells: Autacoids in anti-inflammation. J. Biol. Chem. 278, 14677–14687. http://dx.doi.org/10.1074/jbc.m300218200.
|
| ○ |
Houstis N, Rosen E and Lander E. 2006. Reactive oxygen species have a causal role in multiple forms of insulin resistance.
Nature 440, 944–948. http://dx.doi.org/10.1038/nature04634.
|
| ○ |
Inoue M, Ohtake T, Motomura W, Takahashi N, Hosoki Y, Miyoshi S, Suzuki Y, Saito H, Kohgo Y and Okumura T. 2005. Increased
expression of PPARγ in high fat diet-induced liver steatosis in mice. Bioch and Bioph Res Comms. 336, 215–222. http://dx.doi.org/10.1016/j.bbrc.2005.08.070.
|
| ○ |
Leamy AK, Egnatchik RA and Young JD. 2013. Molecular Mechanisms and the Role of Saturated Fatty Acids in the Progression of
Non-Alcoholic Fatty Liver Disease. Prog Lipid Res. 52:165–174. http://dx.doi.org/10.1016/j.plipres.2012.10.004.
|
| ○ |
Malaguarnera M, Di Rosa M, Nicoletti F, Malaguarnera L. 2009. Molecular mechanisms involved in NAFLD progression. J. Mol. Med. 87, 679–695. http://dx.doi.org/10.1007/s00109-009-0464-1.
|
| ○ |
Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, et al. 2006. International Union of Pharmacology. LXI.
Peroxisome Proliferator-Activated Receptors. Pharmacol Rev. 58, 726–741. http://dx.doi.org/10.1124/pr.58.4.5.
|
| ○ |
Musso G, Gambino G, and Cassader M. 2009. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease
(NAFLD). Prog. in Lipid Research. 48, 1–26. http://dx.doi.org/10.1016/j.plipres.2008.08.001.
|
| ○ |
Nobili V and Sanyal AJ. 2012. Treatment of nonalcoholic fatty liver disease in adults and children: closer look at the arsenal.
J. Gastroenterol. 47, 29–36. http://dx.doi.org/10.1007/s00535-011-0467-x.
|
| ○ |
Poirier Y, Antonenkov VD, Glumoff T and Hiltunen JK. 2006. Peroxisomal β-oxidation: A metabolic pathway with multiple functions.
Mol Cell Res. 1763, 1413–1426. http://dx.doi.org/10.1016/j.bbamcr.2006.08.034.
|
| ○ |
Shapiro H, Tehilla M, Attal-Singer J, Bruck R, Luzzatti R and Singer P. 2011. The therapeutic potential of long-chain omega-3
fatty acids in nonalcoholic fatty liver disease. Clin Nutr. 30, 6–19. http://dx.doi.org/10.1016/j.clnu.2010.06.001.
|
| ○ |
Surwit RS, Kuhn CM, Cochrane C, McCubbin JA and Feinglos MN. 1988. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 37, 1163–1167. http://dx.doi.org/10.2337/diab.37.9.1163.
|
| ○ |
Thanos D, Georgopoulos K, Greenberg M and Leder P. 1988. c-jun dimerizes with itself and with c-fos, forming complexes of
different DNA binding affinities. Cell. 55, 917–924. http://dx.doi.org/10.1016/0092-8674(88)90147-x.
|
| ○ |
Valenzuela R and Videla L. 2011. The importance of the long-chain polyunsaturated fatty acid n-6/n-3 ratio in development
of non-alcoholic fatty liver associated with obesity. Food Funct. 2, 644–648. http://dx.doi.org/10.1039/c1fo10133a.
|
| ○ |
Valenzuela R, Espinosa A, González D, Fernández V, Videla LA, Romanque P, et al. 2012. N-3 long-chain polyunsaturated fatty
acid supplementation significantly reduces liver oxidative stress in high fat induced steatosis. Plos One. 7, e46400. http://dx.doi.org/10.1371/journal.pone.0046400.
|
| ○ |
Videla L and Pettinelli P. 2012. Misregulation of PPAR functioning and its pathogenic consequences associated with nonalcoholic
fatty liver disease in human obesity. PPAR Res. 2012, 1–14. http://dx.doi.org/10.1155/2012/107434.
|
| ○ |
Ye D, Zhang D, Oltman C, Dellsperger K, Lee HC and Van Rollins M. 2002. Cytochrome P-450 epoxygenase metabolites of docosahexaenoate
potently dilate coronary arterioles by activating large-conductance calcium-activated potassium channels. J. Pharmacol. Exp. Ther. 303, 768–776. http://dx.doi.org/10.1124/jpet.303.2.768.
|