Free radical scavenging and α-glucosidase inhibition, two potential mechanisms involved in the anti-diabetic activity of oleanolic acid
J.M. Castellanoa,*, A. Guindaa, L. Macíasa, J.M. Santos-Lozanob,c, J. Lapetrab,d and M. Radaa
aInstituto de la Grasa. CSIC. Food and Health Department. Campus of University Pablo de Olavide. Seville, Spain
bCIBER Physiopathology of Obesity and Nutrition (ciberobn). Instituto de Salud Carlos III. Madrid, Spain
cDepartment of Family Medicine. Primary Care Health District Seville. Primary Care Center San Pablo. Seville, Spain
dDepartment of Family Medicine. Research Unit. Primary Care Health District Seville. Seville, Spain
*Corresponding author: jmcas@ig.csic.es
| |
SUMMARY
This work investigates the role of oleanolic acid (OA), isolated from the olive (Olea europaea L.) leaf, as a radical scavenger and inhibitor of the hydrolyzing enzymes of dietary carbohydrates. New evidence is provided showing that OA may capture 2,2’-azino-bis (3-ethylbenzothiazoline-6-sulphonic acid) and peroxyl radicals, and also exert a strong and non-competitive inhibition of α-glucosidase (IC50 10.11 ± 0.30 μM). The kinetic and spectrometric analyses performed indicate that OA interacts with this enzyme inside a hydrophobic pocket, through an endothermic and non spontaneous process of a hydrophobic nature. These are two possible mechanisms by which OA may facilitate a better control of post-prandial hyperglycaemia and oxidative stress, so contributing to preserving insulin signalling. Obesity, insulin resistance and Type 2 Diabetes Mellitus are considered the first pandemics of the 21st century. In this sense, OA might be used in future preventive and therapeutic strategies, as an ingredient in new drugs and functional foods.
|
| |
RESUMEN
La Captación de radicales libres y la inhibición de α-glucosidasa, dos posibles mecanismos involucrados en la actividad antidiabética del ácido oleanólico. Este trabajo estudia el papel del ácido oleanólico (OA), aislado de la hoja de olivo, como secuestrador de radicales libres
e inhibidor de enzimas implicados en la hidrolisis de los carbohidratos de la dieta, dos mecanismos por los que el triterpeno
podría mitigar la hiperglicemia postprandial y el estrés oxidativo. Se aportan nuevas evidencias que muestran que el OA puede
capturar radicales ácido 2,2’-azino-bis-(3-etilbenzotiazolín)-6-sulfónico y peroxilo, y que ejerce una potente inhibición
no-competitiva de α-glucosidasa (IC50 10.11±0.30 μM). El análisis cinético y espectrométrico llevado a cabo indica que OA interacciona con este enzima en el interior
de un bolsillo hidrofóbico, mediante un proceso endotérmico no espontáneo, de naturaleza hidrofóbica. Estos son dos posibles
mecanismos por los cuales el OA puede facilitar un mejor control de la hiperglucemia postprandial y el estrés oxidativo, lo
que contribuye a preservar la señalización de la insulina. La obesidad, la resistencia a la insulina y la diabetes mellitus
tipo 2 se consideran la primera pandemia del siglo XXI. En este sentido, el OA podría ser utilizado en futuras estrategias
preventivas y terapéuticas, como ingrediente de nuevos fármacos y alimentos funcionales.
|
1. INTRODUCTIONTOP
Type 2 Diabetes Mellitus (T2DM), the most common form of the disease affecting near 90% of diabetic people, is recognized
as a redox disorder, in which oxidative stress is a primary event underlying insulin resistance, β-cell failure, and long-term
diabetes complications (Watson, 2014). This prompted investigations on the therapeutic use of antioxidants, although none of such studies demonstrated clear effects
on the metabolic control of diabetic people (Ceriello and Testa, 2009). Another strategy to minimize oxidative stress is by reducing glycaemia fluctuations during post-prandial periods, since
different studies with cells exposed to levels of glucose elevated intermittently have demonstrated the increased expression
of biomarkers for oxidative stress (Quagliaro et al., 2003). An interesting approach is the inhibition of enzymes involved in carbohydrate breakdown and the intestinal absorption of
glucose. Salivary and pancreatic α-amylases (EC 3.2.1.1) cleaves the α-(1→4) bonds of starch yielding shorter linear and branched
dextrins, which are subsequently hydrolyzed at the non-reducing end into glucose by α-glucosidase (EC 3.2.1.20) in the small-intestinal
brush border membrane. Synthetic inhibitors, such as acarbose, voglibose or miglitol, are widely used in the clinic. However,
the appearance of adverse effects is frequent, including flatulence, abdominal pain, nausea, vomiting, diarrhea, skin hypersensitivity,
an elevation in the hepatic enzymes and even an increased incidence of renal tumors (Fujisawa et al., 2005).
Accordingly, the search for more effective and safer natural bio-molecules, with the dual ability to decrease glucose absorption
and reduce oxidative damage, has become an important scientific goal. Oleanolic acid (OA) (3β-hydroxy-olean-12-en-28-oic acid),
a pentacyclic triterpene widely distributed in the plant kingdom, has attracted the attention of researchers for its interesting
pharmacological properties (Liu, 2005; Dzubak et al., 2006), including anti-diabetic activity (Castellano et al., 2013).
The data available nowadays concerning the antioxidant activity of OA are somewhat contradictory. Its ability to react with
diverse radical species has been evaluated, having obtained an array of varied results (Yin and Chan, 2007; Yang et al., 2007; Wang et al., 2010; Allouche et al., 2011). The OA effect on α-amylase is not completely clear (Khati et al., 2013; Wang et al., 2011). However, a consensus does exist concerning the efficient inhibition of α-glucosidase by the triterpene, and some IC50 values in the micro-molar range have been reported (Ali et al., 2002; Kang et al., 2012). To our knowledge, studies on the mechanism of this reaction have not been carried out. Consequently, in this work, we investigate
the triterpene’s ability to scavenge three radical species (ABTS+, DPPH and peroxyl), and also to inhibit both α-amylase as
α-glucosidase. The paper includes a deeper kinetic study to determine the type of inhibition, as well as fluorescent measurements
to obtain new clues about the mechanism of the OA/α-glucosidase interaction. This research is part of a more general investigation
aimed at exploiting the therapeutic potential of OA against diabetes.
2. MATERIALS AND METHODSTOP
2.1. Plant material, reagents and standardsTOP
Olive (Olea europaea L. cv. Picual) leaves were hand-picked from adult trees (>10 years old) from the experimental olive grove belonging to the
Instituto de la Grasa-CSIC in Seville.
All solvents used were of analytical grade. Betulinic acid; hexamethyldisilazane; trimethylchlorosilane; 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic
acid) (ABTS); 2,2-Diphenyl-1-picrylhydrazyl (DPPH); 2,2’-azobis (2-amidinopropane) dihydrochloride (AAPH); fluorescein; pancreatic
α-amylase; potato starch; 3,5-dinitrosalicylic acid; sodium potassium tartrate; α-glucosidase from Saccharomyces cerevisiae; 4-nitrophenyl-α-D-glucopyranoside (p-NPG); and acarbose were purchased from Sigma-Aldrich (Sigma-Aldrich Quimica, Madrid, Spain).
2.2. Isolation of oleanolic acid from O. europaeaTOP
High purity OA was isolated from olive leaves according to the procedure described by us (Guinda et al., Patent 2001/2160553). Briefly, dry leaves were extracted by maceration with 96% ethanol, which was then percolated and subsequently reduced by vacuum. Solid OA was obtained by crystallization and filtration. Its chemical nature was confirmed by GC-MS (P Gutiérrez-Adánez, Thesis of Master, University Pablo de Olavide; Seville, 2013), and its purity was determined by differential scanning calorimetry (DSC) and GC-FID (Albi et al., 2001; Guinda et al., 2010).
2.3. Derivatization of oleanolic acidTOP
The low volatility and high molecular weight of OA demands its derivatization prior to the GC analysis. The silylating reagent
was prepared by mixing 3 mL of hexamethyldisilazane and 1 mL of trimethylchlorosilane with 9 mL of anhydrous pyridine. Ten
milligrams of OA were dissolved in 20 mL absolute ethanol. An aliquot (100 μL) of this solution and 100 μL of 0.5 mg/mL betulinic
acid (internal standard) were placed in 1 mL gas-tight vials and evaporated to dryness under a N2 stream. The residue was dissolved in 200 μL of the silylating reagent and sonicated at 70 °C for 15 min. The solution harboring
the derivatized triterpene was analyzed by GC-FID and GC-MS.
2.4. GC-FID AnalysisTOP
The quantification of OA was carried out by a modification of the method described by us in (Guinda et al., 2010). Concisely, 1 μL of the silylated sample was injected into an Agilent 6890N GC (Agilent Technologies, CA), equipped with
a Rtx-65TG Cross-bond capillary column (30 m × 0.25 mm I.D.; 0.1 mm film thickness) coated with 35% dimethyl-65% diphenyl
polysiloxane as stationary phase (Restek, Co., Bellefonte, PA) and a FID detector. The injection was done in the split mode,
and hydrogen was used as carrier gas (pressure at column head 140 kPa). The oven temperature was maintained at 260 °C, and
the injector and detector temperatures at 300 °C.
2.5. GC-MS identification of oleanolic acidTOP
The analysis of derivatized OA was performed using a coupled gas chromatograph-mass spectrometry detector (GC-MS) QP2010 Ultra
(Shimadzu Europa GmbH) fitted with an AOC-20i auto-sampler, an ion source of electron impact and a quadrupole detector. The
splitless mode was used and the injector temperature was set at 290 °C. Helium was the carrier gas at a pressure of 53.1 kPa
and a flow of 1 mL/min. The oven temperature program was as follow: initial temperature 50 °C for 1 min, 50–200 °C at 40 °C/min,
200–280 °C at 10 °C/min, held at 280 °C for 8.50 min, and finally at 300 °C for 1 min. Total run time: 24,25 min. The MS conditions
were: interface temperature: 280 °C; ion source temperature: 220 °C; electron impact: 70 eV; acquisition mode: scan (m/z 50–600).
The identification of the OA trimethylsilyl derivative was accomplished by comparing the retention time and abundance ratio
of two fragment ions (203 and 189 m/z) with those of the standard compound.
2.6. Purity evaluation by DSCTOP
The OA purity was also assessed by Differential Scanning Calorimetry (DSC), following the method described in (Albi et al., 2001). A TA-Q2000 calorimeter equipped with a cooling unit (Thermal Analysis Instruments; Barcelona, Spain) was used. This instrument
was calibrated with indium (99.9 %). The OA samples (10–15 mg) were enclosed in hermetically sealed aluminum pans and analyzed
against air (empty pans) as reference. The calorimeter chamber was previously thermostatized at 30 °C for 10 min. Then, the
melting curves from 30 to 350 °C (heating rate 10 °C/min) were recorded. The data were analyzed using the Peak Program software
of the TA-Q2000.
2.7. Radical scavengingTOP
2.7.1. 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) assayTOP
The ABTS+ scavenging capacity was evaluated following a modification of the procedure by Re et al. (1999). The radicals were generated by oxidizing its precursor ABTS (7.0 mM in methanol) with potassium persulfate (2.45 mM), maintaining the mixture overnight in the dark at room temperature. The resultant ABTS+ solution was diluted to have an absorbance at 734
nm in the range of 0.7–0.8. An aliquot (10 μL) of OA samples (0.5, 1.0, 4.0 and 10.0 mM in methanol), or a blank (methanol),
was added to 1 mL of the ABTS+ solution. The mixture was stirred and left to react in the dark at room temperature for 5 min,
and then the absorbance at 734 nm was recorded in a Beckman DU 640 UV-VIS light spectrophotometer (Beckman Coulter Inc.; Pasadena,
CA; USA). The results were expressed as percentage of the initial absorbance of the ABTS+ solution. Butylated hydroxytoluene
(BHT) (0.5 mM in methanol) was assayed as reference antioxidant.
2.7.2. 2,2-Diphenyl-1-picrylhydrazyl (DPPH) free radical scavenging testTOP
The OA ability to capture DPPH radicals was also assessed spectrophotometrically according to the revised protocol by Sharma and Bhat (2009). The reaction mixture (4 mL), containing 50 μM DPPH alone or in the presence of OA (1, 25, 50, 100 and 1000 μM), was incubated
for 30 min at 30 °C in the dark, and then the absorbance at 517 nm was measured using the Beckman DU 640 spectrophotometer.
The scavenging activity was expressed as percentage of the initial absorbance of the DPPH solution. BHT (50 and 100 μM) was
also included in the assay as a positive control.
2.7.3. Oxygen Radical Absorbance Capacity (ORAC) AssayTOP
The ORAC assay was performed following a modification of the procedure reported by Prior et al. (2003), in which fluorescein was the target of the peroxyls generated with 2,2’-azobis (2-amidinopropane) dihydrochloride (AAPH).
An aliquot (20 μL) of the OA sample [0.1–0.7 mM in 10% DMSO (in 75 mM phosphate buffer, pH 7.4 at 37 °C)] or a blank (buffered
DMSO) was pre-incubated with 120 μL of 116.7 nM fluorescein for 10 min at 37 °C. Then, the reaction was started by the addition
of 60 μL of 40 mM AAPH. The time course of the fluorescent decay (λex = 485 nm, λem = 525 nm) was recorded for 80 min using
a Fluostar Galaxy plate reader (BMG Labtechnologies, Offenburg, Germany). The capacity to scavenge peroxyl radicals correlates
with the net area under the fluorescent decay curve (AUC), calculated by subtracting the OA sample AUC from the blank AUC.
The AUCs were automatically calculated by the Fluostar software. The ORAC value (expressed as the μmol Trolox equivalent),
was determined from the calibration curve obtained with standard Trolox in the concentration range 1–10 μM.
2.7.4. Rancimat methodTOP
Rancimat 743 equipment (Metrohm AG; Erisau, Switzerland) was used for evaluating the role of OA against lipid peroxidation.
An aliquot (300 μL) of OA solutions (0.5 and 1.0 mM in DMSO) was added to 3.0 g of olive or sunflower oil. The mixture was
heated at 100 °C whereas an air flow (20 L/h) bubbled constantly. The effluent gas was collected in de-ionized water, whose
electrical conductivity was continuously monitored. In this system, the antioxidant activity correlates with the extension
of timing up to the inflection point in the conductivity curve (induction time) (Burkow et al., 1995). The antioxidant activity index (AAI) was calculated from the ratio induction time of the oil with OA/induction time of oil alone. BHT (250 and 500 μg/g) was likewise included in the assays as a reference antioxidant.
2.8. In vitro inhibition of carbohydrate hydrolyzing enzymesTOP
2.8.1. In vitro assay for α-amylaseTOP
α-Amylase was assayed using the chromogenic method adopted by Sigma–Aldrich (www.sigmaaldrich.com/technical-documents/protocols/biology/enzymatic-assay-of-a-amylase.html). Porcine pancreatic α-amylase was dissolved in an ice-cold 20 mM sodium phosphate buffer (pH 6.9 at 37 °C) containing 6.7
mM NaCl. The potato starch solution (1% w/v) was prepared in the same buffer.
In a first series of experiments, 40 μL of OA solutions (5–1000 μM in buffered 10% DMSO), 60 μL of phosphate buffer, and 400
μL of the substrate solution were mixed in a screw-top glass tube, and pre-incubated at 37 °C for 5 min. The reaction was
subsequently started by the addition of 200 μL of the enzyme solution (1 unit/mL), and the mixture was incubated at the same
temperature for 5 min. Then, 350 μL of the DNS color reagent (96 mM 3,5-dinitrosalicylic acid, 5.31 M sodium potassium tartrate
in 2 M NaOH) were added to the tube and placed into a 85 °C water bath for 15 min. Then, the mixture was removed and diluted
with 3 mL distilled water. The maltose generated in the reaction (absorbance at 540 nm) was quantified from the calibration
curve obtained with standard maltose in the range of 50 μM–3.75 mM (A540 = 2.6185 ∙ 10–4 [maltose] – 0.0279; r2 = 0.9972). Control incubations representing 100% enzyme activity were conducted in an identical fashion, replacing the OA
solution by DMSO. Blank incubations, where the enzyme solution was replaced with phosphate buffer, were also included. In
a second series of assays, the enzyme was pre-incubated with OA at 37 °C, and the reaction was started by the addition of
the starch solution. In both series, acarbose (20 μM) was introduced as a reference inhibitor.
2.8.2. In vitro assay for α-glucosidaseTOP
The α-glucosidase activity was tested by adapting the protocol by Kang et al. (2011) to 96-well micro-plates. To each well, 30 μL of a 100 mM phosphate buffer containing 100 mM NaCl (pH 6.8 at 37 °C), 30 μL
of 2.5 mM p-NPG in this same buffer, and 30 μL of OA (0.05–50.0 μM in buffered 10% DMSO), were added. The plate was pre-incubated at
37 °C for 5 min, and the reaction was started by adding 30 μL of the α-glucosidase solution (0.2 U/mL α-glucosidase in phosphate
buffer). After 20 min at the same temperature, 120 μL of 100 mM sodium carbonate were added to stop the reaction. The release
of 4-nitrophenol (ε = 18.3 mM−1 cm−1) was determined by measuring the absorbance at 405 nm in a Multiskan Spectrum (Thermo Labsystems; Milford, MA, USA) micro-plate
reader. One enzyme activity unit is defined by the release of 1.0 μmol α-D-glucopyranoside per minute at 37 °C. Control incubations
representing 100% enzyme activity were conducted, replacing OA solutions with DMSO. Likewise, blank incubations were carried
out where the enzyme solution was replaced by the phosphate buffer. The inhibitory activity of OA was assayed in the range
of 0.05–50.0 μM, and 20 μM acarbose was introduced as reference inhibitor.
2.8.3. Fluorescence quenching measurementsTOP
In these experiments, α-glucosidase was assayed at 1 μM in the presence of OA (0–25 μM). The reaction mixture (whose final
volume was brought to 4.0 mL with phosphate buffer) was incubated for 10 min at 20, 30 and 37 °C. The fluorescent spectra
of α-glucosidase were obtained on a Cary Eclipse fluorescence spectrophotometer (Agilent Technologies; Santa Clara, CA, USA)
equipped with a 10.0 mm quartz cell and a thermo-stated bath. The excitation wavelength was 280 nm, and the emission spectra
were recorded from 300 to 500 nm. The excitation and emission bandwidths were both set at 5 nm. Appropriate blanks corresponding
to the phosphate buffer alone were assayed and subtracted to correct background fluorescence.
2.9. Statistical analysisTOP
All data are presented as the means ± standard deviation (SD) from a series of three independent experiments carried out with
five replicates. The data were evaluated by a one-way ANOVA with the SigmaPlot 12.5 software (SPSS, Inc., Chicago, IL), and
the differences between means were assessed using the Duncan’s multiple-range test. Statistical significance was considered
at p < 0.05.
3. RESULTSTOP
3.1. Obtaining pure oleanolic acid from olive leaf title OneTOP
Eighteen grams and six hundred thirty two milligrams of crystallized OA were obtained from one kilogram of dried olive leaves.
Its purity (98.6%) was determined by both GC-FID and DSC (data not shown).
3.2. Free radical scavengingTOP
3.2.1. 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS+) assayTOP
The stable radical ABTS+ is commonly used for the estimation of total antioxidant capacity. The simpler and more frequently
applied approach of this assay consists of analyzing the activity of a sample against pre-formed radicals. With this scheme,
we have found that OA is a moderate scavenger of ABTS+, and its action is dose-dependent (Table 1). Thus, 10 mM OA removed 24% of the initially present ABTS+. In comparison, 0.5 mM BHT abolished these radicals almost completely.
On the other hand, we have tested two other solvents in addition to methanol, for preparing the OA solutions: carboxymethylcellulose
(CMC) and DMSO, but they did not improve the scavenging activity of the triterpene.
Table 1. Scavenging of the ABTS+ radical by oleanolic acid. Results are expressed as the means ± SD (n = 15). CMC = carboxymethylcellulose [2.5% (w/w)]. BHT was included in the assay as positive control. Different letters within a column indicate that values are different at p<0.05
|
absorbance (λ734)
|
reduction (%) |
| ABTS+ |
0.779 ± 0.041a |
|
| + OA (mM) in methanol |
|
|
| 0.5 |
0.682 ± 0.035b,d |
12.5 ± 0.6 |
| 1.0 |
0.676 ± 0.038b,d |
13.2 ± 0.7 |
| 4.0 |
0.645 ± 0.047b,e |
17.2 ± 1.2 |
| 10.0 |
0.586 ± 0.030c,h |
24.8 ± 1.3 |
| + OA (mM) in CMC |
|
|
| 0.5 |
0.691 ± 0.054d |
11.2 ± 0.9 |
| 1.0 |
0.673 ± 0.034b,d,f |
13.5 ± 0.7 |
| 4.0 |
0.636 ± 0.032e,f,h |
18.4 ± 0.9 |
| + OA (mM) in DMSO |
|
|
| 4.0 |
0.631 ± 0.028e,g,h |
18.9 ± 0.9 |
| 10.0 |
0.599 ± 0.031h |
23.2 ± 1.2 |
| + BHT 0.5 mM |
0.026 ± 0.002i |
96.7 ± 3.5 |
3.2.2. 2,2-Diphenyl-1-picrylhydrazyl (DPPH) free radical scavenging testTOP
DPPH is also very common for the study of natural antioxidants. Its solutions have a characteristic deep purple color (λmax
515–517 nm) that decolorize by the action of radical scavengers. Performing this procedure, it became evident that OA does
not capture DPPH radicals, at least at concentrations lower than 1 mM (data not shown). The use of DMSO substituting for methanol
as the triterpene solvent did not provide better results.
3.2.3. Oxygen Radical Absorbance Capacity (ORAC) AssayTOP
The ORAC assay determines the ability of a compound to preserve fluorescein from the attack of peroxyls generated with AAPH.
Using this method, OA showed a low and dose-dependent capability to react with these radicals (Figure 1). The net areas under the fluorescent decay curves were linearly fitted to the OA concentrations in the range of 0.1–0.7
mM. An ORAC value of 1.628 nmol Trolox equiv/μmol OA was determined from the calibration curve obtained with standard Trolox
(Net AUC = 19.904 nmol Trolox + 6.335; r2 = 0.9899).
|
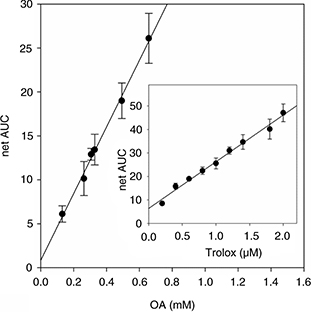 Figure 1. Oxygen radical absorbance capacity (ORAC) assay for OA. Net area under the fluorescent decay curve in the presence of increasing
concentrations of OA. The inset is the calibration curve obtained with standard Trolox. Results are expressed as mean ± SD
of three independent experiments carried out with five replicates. Figure 1. Oxygen radical absorbance capacity (ORAC) assay for OA. Net area under the fluorescent decay curve in the presence of increasing
concentrations of OA. The inset is the calibration curve obtained with standard Trolox. Results are expressed as mean ± SD
of three independent experiments carried out with five replicates.
|
|
3.2.4. Rancimat methodTOP
The Rancimat method, based on the induction of lipid oxidation by exposure to airflow at high temperatures, is likely the
most commonly used procedure for measuring the oxidative stability of oils and fats. The process occurs via a chain reaction
(with participation of hydroxyl and peroxyl radicals), in which the double bonds of unsaturated fatty acids undergo cleavage,
releasing volatile short-chain organic acids, aldehydes and ketones. The reaction is characterized by a relatively slow initial
period before it suddenly speeds up. The time for this to happen, called induction time, is taken as an indication of the
oxidative stability of the lipid sample. The Rancimat procedure can be also applied for assessing the efficiency of antioxidants,
by appraising how long they are able to prolong the initial induction step. This paper reports the use of such equipment for
evaluating the antioxidant potential of OA for the first time.
We first ran experiments with an olive oil, chosen by its low content in minor components. In this lipid system, 0.5 mM OA
did not prevent oil oxidation, but instead it exerted a slight pro-oxidant activity, as evidenced by the shortening of the
induction period (Figure 2). By doubling the OA concentration in the medium no significant changes were observed. A second sequence of trials was performed
with refined sunflower oil, whose triglycerides are richer in polyunsaturated fatty acids than those of olive oil. In that
lipid matrix, the triterpene showed a clear and dose-dependent pro-oxidant action. So, in the presence of 1.0 mM OA the oxidative
stability of the sunflower oil fell by 50%. In both experimental series, BHT was able to remarkably delay oil oxidation. To
the best of our knowledge, this is the first time a pro-oxidant action of OA is reported.
|
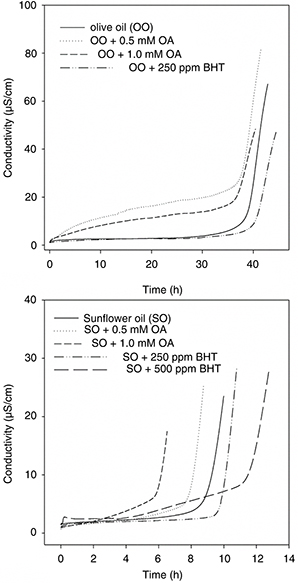 Figure 2. Effect of OA on the oxidative stability of olive and sunflower oils (Rancimat method). Typical conductivity vs time curves representative of three independent experiments performed with five replicates. Figure 2. Effect of OA on the oxidative stability of olive and sunflower oils (Rancimat method). Typical conductivity vs time curves representative of three independent experiments performed with five replicates.
|
|
3.3. Inhibition of carbohydrate hydrolyzing enzymesTOP
3.3.1. Effect of OA on the pancreatic α-amylaseTOP
α-Amylase activity was assayed using potato starch as substrate to determine the maltose produced in the reaction. We performed
two series of experiments, considering whether starch and OA were first pre-incubated and the reaction was started by the
addition of the enzyme solution; or whether the enzyme was first pre-incubated with the triterpene and then the reaction initiated
by addition of the substrate. With both approaches, no enzyme inhibition was observed in the OA concentration range 5.0 μM–1.0
mM. However, 20 μM acarbose (the reference inhibitor) was able to repress the enzyme activity by 27.2 and 30.1%, respectively
(data not shown).
3.3.2. Inhibition of the α-glucosidase activity by OATOP
Unlike α-amylase, OA caused a strong inhibition of the α-glucosidase activity. This effect was dose-dependent, with the enzyme
activity decreasing remarkably when the OA dose augmented (Figure 3). In fact, the α-glucosidase action was virtually abolished at 20 μM OA and above. The dose resulting in 50% inhibition (IC50) was estimated to be 10.11 ± 0.30 μM.
|
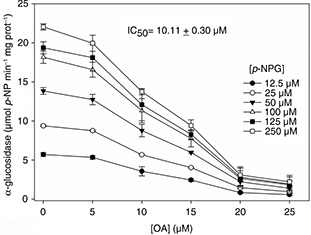 Figure 3. Inhibitory effect of OA on the α-glucosidase activity. In the inset the OA dose resulting in 50% inhibition (IC50) is expressed. Results are expressed as mean ± SD of three independent experiments carried out with five replicates. Figure 3. Inhibitory effect of OA on the α-glucosidase activity. In the inset the OA dose resulting in 50% inhibition (IC50) is expressed. Results are expressed as mean ± SD of three independent experiments carried out with five replicates.
|
|
For a deeper study of the inhibition mechanism we first corroborated that α-glucosidase catalysis followed a Michaelis-Menten
kinetic. The reaction proceeded at its maximum rate (26.84 μmol p-NP min−1 mg prot−1) with substrate concentrations greater than 150 μM, and a Km value of 51.34 μM was determined for p-NPG. We also studied the time-course of the reaction in the absence and presence of OA (5, 15 and 25 μM), finding that the
activity vs time relationship progressed linearly for at least 25 min (data not shown). Then, we evaluated the enzyme activity at increasing
concentrations of p-NPG, in the presence of different doses of OA. The Lineweaver-Burk’s kinetic analysis of results indicated that OA was a
non-competitive inhibitor of α-glucosidase (Figure 4). The inhibition constant (Ki) of 9.12 ± 0.24 μM was calculated by both the Dixon and Cornish-Bowden analyses.
|
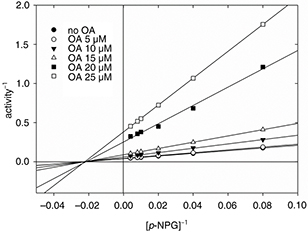 Figure 4. Lineweaver-Burk plots for the α-glucosidase inhibition by OA. Figure 4. Lineweaver-Burk plots for the α-glucosidase inhibition by OA.
|
|
3.3.3. Fluorescence quenching of α-glucosidase by oleanolic acidTOP
The powerful inhibition that OA exerts on α-glucosidase suggests that the triterpene directly binds to the protein. To achieve
new evidence for this interaction, we conducted, for the first time to our knowledge, binding studies employing fluorescent
spectroscopy. The spectra of α-glucosidase in the presence of different OA concentrations were recorded at 20, 30 and 37 °C
(Figure 5). Under the same experimental conditions, OA did not display intrinsic fluorescence or UV-VIS absorption in the wavelength
range 300–500 nm, not interfering with the α-glucosidase spectra (data not shown). α-Glucosidase exhibited a strong fluorescent
emission peak at 333 nm after excitation at 280 nm, indicating that tryptophans located in inner regions of the protein are
practically the only ones emitting fluorescence. In the presence of OA, significant and dose-dependent decreases in the fluorescent
intensity were observed. This quenching effect did not involve hypsochromic or bathochromic shifts, which reveals that the
OA/α-glucosidase interaction only affected those tryptophyls positioned within a cleft of the enzyme.
|
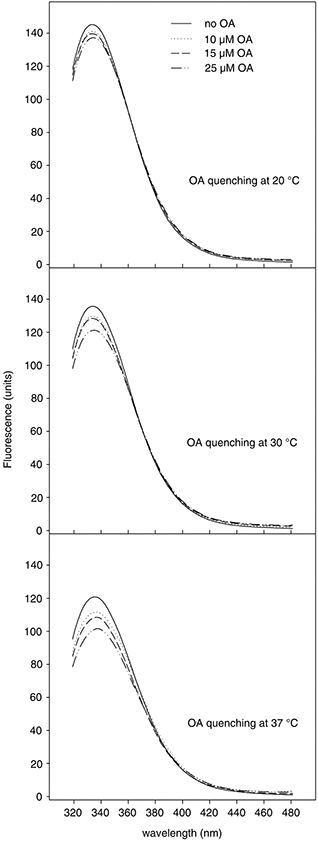 Figure 5. Fluorescent emission spectra (300–500 nm) of α-glucosidase at 20, 30 and 37 °C, in the presence of increasing concentrations
of OA (λex = 280 nm). Figure 5. Fluorescent emission spectra (300–500 nm) of α-glucosidase at 20, 30 and 37 °C, in the presence of increasing concentrations
of OA (λex = 280 nm).
|
|
Figure 6 shows the Stern-Volmer plots (F0/F = 1 + KSV[OA]) for the quenching of α-glucosidase fluorescence by OA. In this equation, F0 and F denote the steady-state fluorescent intensities in the absence and presence of the triterpene, respectively; and KSV is the Stern-Volmer quenching constant, a measure of the interaction strength between the protein and its ligand. The plot’s
linearity illustrates that within the assayed concentration range, a single class of enzyme fluorophores is affected by OA.
The results also show that the KSV values increased with increasing temperature, suggesting that quenching proceeded mainly through a dynamic mechanism of collisions
between OA and tryptophans. This relationship may also be described by the equation log [(Fo- F)/ F] = log KA+ n log [Q] (Yan et al., 2014), where KA is the binding constant, and n the number of binding sites on the protein (Figure 6). It can be observed that both KA and n rose with the increase in temperature, showing again that the OA/α-glucosidase interaction was favored under warmer conditions.
Moreover, the values of n show a trend to one, supporting the existence of a single class of binding site for OA.
|
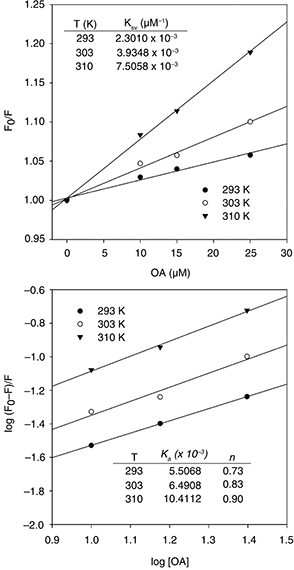 Figure 6. Stern-Volmer plots for the fluorescence quenching of α-glucosidase by OA. Figure 6. Stern-Volmer plots for the fluorescence quenching of α-glucosidase by OA.
|
|
More information about this interaction may be inferred from the signs of the thermodynamic parameters in the process. The changes in enthalpy (ΔH) and entropy (ΔS) can be determined from the Van’t Hoff equation [ln KSV = - (ΔH/RT) + (ΔS/R); where R is the gas constant], and the free energy changes (ΔG) can be estimated from the ΔG=ΔH–TΔS relationship. Both ΔH and ΔS were positive, proposing that the OA/α-glucosidase association has a hydrophobic nature. Likewise, ΔG values were positive, signifying that the triterpene/protein association is endergonic in the assayed temperature range
(20–37 °C).
4. DISCUSSIONTOP
The experimental and clinical evidence indicates an inverse association between insulin sensitivity and oxidative stress,
up to the point that diabetes is acknowledged as a phenotype of dysfunctions in mitochondria and endoplasmic reticulum, associated
with an excessive ROS production or an impairment of the endogenous antioxidant system (Lowell and Shulman, 2005). Because post-prandial hyperglycaemia induces oxidative stress, the search for natural bio-molecules with the dual ability
to decrease glucose absorption and oxidative damage has become an important research topic. Within that scenario, this paper
reports new findings about OA as a radical scavenger and inhibitor of the α-glucosidase activity.
The antioxidant power of OA is subject to controversy since it is markedly influenced by the singularities of the experimental
systems employed in its evaluation. We have evaluated the OA ability to scavenge three radical species (ABTS+, DPPH and peroxyl),
finding that it can capture ABTS+ radicals in a moderate and dose-dependent manner, but it lacks the ability to scavenge DPPH
species. These results are in agreement with those previously provided by other authors (Yang et al., 2007; Wang et al., 2010; Allouche et al., 2011).
The role of triterpene against peroxyls deserves a more detailed discussion. The ORAC assay has demonstrated that OA moderately
captures the radicals generated from AAPH, although it failed with those formed from complex lipid matrixes in the Rancimat
procedure. In fact, this last protocol has revealed that OA might exert a pro-oxidant action. These results do not coincide
with those of Yin and Chan (2007), who have studied the triterpene potential for reducing peroxyl radicals using a modified TBARS assay on liposomes. They
determined that OA had a high and dose-dependent activity when liposome peroxidation was induced by AAPH and 2,2’-azobis-(2,4-dimethylvaleronitrile).
Closer to the Rancimat method, Assimopoulou et al. (2005) investigated the OA effect on the oxidation of lard and sunflower oil in an oven at 65 °C. They monitored the evolution
of peroxide value, reporting that OA did not exhibit a clear antioxidant activity. Wang et al. (2010) examined the effect of OA in liver microsomes of Sprague-Dawley rats, comparing the triterpene action with those of well-known
antioxidants, and found that the protection offered by OA against peroxyls depended on the substance used for inducing lipid
peroxidation. This study also evidenced that some molecules recognized as antioxidants (vitamin C or α-lipoic acid) could
act as pro-oxidants depending of the nature of the lipid oxidizing agent. Once again, different experimental approaches lead
to different conclusions, many of which are conflicting.
Another mechanism by which OA may contribute to controlling oxidative stress is by decreasing post-prandial hyperglycaemia,
through the inhibition of carbohydrate-hydrolyzing enzymes. In this paper, we present results proving that OA is not an α-amylase
inhibitor. This finding is in agreement with other published data (Hou et al., 2009; Wang et al., 2011) although there are also contradictory results supporting that OA might effectively repress this activity (Khati et al., 2013; Ali et al., 2006; Komaki et al., 2003), with quite variable IC50 values in the range of 10 μM–2.6 mM.
Unlike α-amylase, OA exerts a strong and dose-dependent inhibition of α-glucosidase. We have determined an IC50 10.11 ± 0.30 μM, a very similar value to those reported by other authors (Ali et al., 2002; Hou et al., 2009; Kang et al., 2012). In addition, we present here new contributions to the understanding of the OA/α-glucosidase interaction, performing a deeper
kinetic study and fluorescent measurements. We found that OA is a non-competitive inhibitor of α-glucosidase (Ki = 9.12 ±
0.24 μM), which binds the enzyme in a neighboring region of the active site. The comparison of the inhibitory efficiency of
OA with those of other natural and synthetic structural analogs allows us to outline some molecular features with impact on
inhibition: a) the relevancy of the hydrophobic skeleton with a hydroxyl group in C3 and a carboxyl group in C28 is acknowledged;
b) the introduction of a second phenolic hydroxyl at C2, as in maslinic or corosolic acids, might increase the inhibitory
power of the triterpene (Ali et al., 2006); c) the substitution of the methyl at C23 by a more bulky ligand (an acetyl group, for example) would diminish this ability
(Ali et al., 2002); and d) the β-orientation of the phenolic hydroxyl in C3 seems to be clearly influential for the inhibition, since corosolic
acid with a β-oriented hydroxyl such as OA exhibits an IC50 value of 7.47 μM45, whereas 3-epi-corosolic acid, its α-stereoisomer, displays a IC50 63.9 μM (Chowdhury et al., 2014).
The fluorescent measurements of the OA/α-glucosidase complex provide useful information about the binding mechanism and binding
site. The intrinsic fluorescence of a folded protein is the result of that emitted by its individual aromatic residues, with
most of the emissions due to the excitation of tryptophans and few due to tyrosines and phenylalanines. On the other hand,
tryptophans located in inner regions of proteins with hydrophobic microenvironments emit maximum fluorescence near 331 nm,
whereas those residues located on the protein surface exposed to a high-polarity aqueous media emit at longer wavelengths
(≈ 350 nm) (Lacovicz, 2006). Therefore, the wavelength of maximum emission can be interpreted in terms of the ratio between these two classes of tryptophans.
Moreover, intrinsic fluorescence often decreases when a protein interacts with another compound. The process (quenching) happens
during the excited state lifetime (collisional or dynamic quenching) or due to the formation of a complex in the ground state
(static quenching).
In our case, the maximum fluorescence emitted by α-glucosidase as well as quenching by interaction with OA are detected at
333 nm, indicating that the tryptophyls involved in the triterpene binding are almost exclusively located inside a hydrophobic
pocket. The Stern-Volmer plots determined a single class of binding site on α-glucosidase (n tending towards 1), with all the participating tryptophans identically accessible (linearity of the plots within the experimental
range of OA concentrations). We have also found that the α-glucosidase/triterpene interaction responds to a collisional mechanism
which is favored at warmer temperatures, such as that operating in the human body (rising values of quenching constant at
increasing temperature). The study of thermodynamic parameters corroborated the idea that inhibition of α-glucosidase by OA
is an endothermic and entropy-driven, a non-spontaneous process of predominant hydrophobic nature. However, complementary
contribution of hydrogen bridges cannot be ruled out because the reaction medium is aqueous and both OA and α-glucosidase
molecules include prominent hydroxyl groups. These outcomes are coherent with reported data postulating the existence of tryptophans
neighboring the α-glucosidase active site, which are likely to be more relevant for maintaining an efficient substrate accommodation
rather than for the catalysis itself (Faridmoayer and Scaman, 2005). So, the OA binding to these tryptophyls would alter the folding of the protein, hindering the access of the substrate to
the inner cleft with the active site.
In addition to studies on the inhibition of amylase and glucosidase activities, in recent years new data have been reported
showing that OA can also repress the expression of these proteins in the small intestine (Gabás-Rivera et al., 2013; Khathi et al., 2013), in addition to the glucose transporters SGLT1 and GLUT2, involved in the absorption of the saccharide from the lumen of
the small intestine to the bloodstream.
In summary, our results support the hypothesis that OA may facilitate a better control of postprandial hyperglycaemia and
oxidative stress, so contributing to the preservation of insulin signalling. While its aptitude for inhibiting α-glucosidase
is powerful, its ability for acting as scavenger of radical species is only moderate. This last aspect indicates that the
efficient antioxidant activity exhibited by OA in cell cultures and experimental animals is likely to result from a more intricate
and multi-factorial mechanism. In recent years, there have been increasing evidence to support the assertion that OA reinforces
the cellular antioxidant response by modulating the expression of key genes, including antioxidant proteins and NADPH-producing
enzymes, responsible for maintaining the GSH level and its transportation into mitochondria. In this nutri genomic effect
of OA, the activation of the nuclear factor erythroid 2 p45-related factor 2 (Nrf2) seems to play a protagonist role (Wang et al., 2010; Castellano et al., 2013).
Obesity, insulin resistance and T2DM are considered the first pandemics of the 21st century. Therefore, National Health Systems
are obliged to introduce urgent measures which delay or avoid their progression. In this sense, OA might be used in future
preventive and therapeutic strategies. Despite the limited number of studies in humans, it is accepted that OA is a safe phytochemical.
None of these trials have recorded significant adverse effects (Xu, 1980; Minich et al., 2007; Song et al., 2006; Chen et al., 2010; Rada et al., 2015). Nevertheless, further investigations in humans will be necessary to extend the use of OA in the design of new drugs and
functional foods, which allow for personalized diets and nutri-genomic approaches for the prevention of high-prevalence chronic
disorders such as diabetes.
ACKNOWLEDGMENTSTOP
The Spanish Ministry of Economy and Competitiveness (Carlos III Institute of Health) financed the research project PI10/01415.
The authors wish to thank the Andalusian Public System of Health (Spain) and the ACESUR Group for making the PREDIABOLE Study
possible.
REFERENCESTOP
| ○ |
Albi T, Guinda A, Lanzón A. 2001. Procedimiento de obtención y determinación de ácidos terpénicos de la hoja de olivo (Olea europaea). Grasas Aceites 52, 275–278.
|
| ○ |
Ali H, Houghton PJ, Soumyanath A. 2006. α-Amylase inhibitory activity of some Malaysian plants used to treat diabetes; with
particular reference to Phyllanthus amarus. J. Ethnopharmacology 107, 449–455. http://dx.doi.org/10.1016/j.jep.2006.04.004 |
| ○ |
Ali MS, Jahangir M, Hussan SS, Choudhary MI. 2002. Inhibition of α-glucosidase by oleanolic acid and its synthetic derivatives.
Phytochemistry 60, 295–299. http://dx.doi.org/10.1016/S0031-9422(02)00104-8 |
| ○ |
Allouche Y, Warleta F, Campos M, Sánchez-Quesada C, Uceda M, Beltrán G, Gaforio JJ. 2011. Antioxidant, antiproliferative and
pro-apoptotic capacities of pentacyclic triterpenes found in the skin of olives on MCF-7 human breast cancer cells and their
effects on DNA damage. J. Agric. Food Chem. 59, 121–130. http://dx.doi.org/10.1021/jf102319y |
| ○ |
Assimopoulou AN, Zlatanos SN, Papageorgiu VP. 2005. Antioxidant activity of natural resins and bioactive triterpenes in oil
substrates. Food Chem. 92, 721–727. http://dx.doi.org/10.1016/j.foodchem.2004.08.033 |
| ○ |
Brown BG, Zhao XQ, Chait A, Fischer LD, Cheung MC, Morse JS, Dowdy AA, Marino EK, Bolson EL, Alaupovic P, Frohlich J, Albers
JJ. 2001. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N. Eng. J. Med. 345, 1583–1592. http://dx.doi.org/10.1056/NEJMoa011090 |
| ○ |
Castellano JM, Guinda A, Delgado T, Rada M, Cayuela JA. 2013. Biochemical basis of the antidiabetic activity of oleanolic
acid and related pentacyclic triterpenes. Diabetes 62, 1791–1799. http://dx.doi.org/10.2337/db12-1215 |
| ○ |
Ceriello, A.; Testa, R. 2009. Antioxidant anti-inflammatory treatment in type 2 diabetes. Diabetes Care 32, S232–S236. http://dx.doi.org/10.2337/dc09-S316 |
| ○ |
Chen RJ, Liu X, Li PM, Zhang L, Zhao L and Zhang XL. 2010. Pharmacokinetic profiles of oleanolic acid formulations in healthy
Chinese male volunteers. Chinese Pharmaceutical J. 45, 621–626.
|
| ○ |
Chowdhury SS, Islam MN, Jung HA, Choi JS. 2014. In vitro antidiabetic potential of the fruits of Crataegus pinnatifida. Res. Pharm. Sci. 9, 11–22.
|
| ○ |
Dzubak P, Hajduch M, Vydra D, Hustova A, Kvasnica M, Biedermann D, Markova L, Urban M, Sarek J. 2006. Pharmacological activities
of natural triterpenoids and their therapeuric implications. Nat. Prod. Rep. 23, 394–411. http://dx.doi.org/10.1039/b515312n |
| ○ |
Faridmoayer A, Scaman CH. 2005. Binding residues and catalytic domain of soluble Saccharomyces cerevisiae processing alpha-glucosidase I. Glycobiology 15, 1341–1348. http://dx.doi.org/10.1093/glycob/cwj009 |
| ○ |
Fujisawa T, Ikegami H, Inoue K, Kawabata Y, Ogihara T. 2005. Effect of two α-glucosidase inhibitors, voglibose and acarbose,
on postprandial hyperglycemia correlates with subjective abdominal symptoms. Metabolism 54, 387–390. http://dx.doi.org/10.1016/j.metabol.2004.10.004 |
| ○ |
Gabás-Rivera C, Martínez-Beamonte R, Ríos JL, Navarro MA, Surra JC, Arnal C, Rodríguez-Yoldi MJ, Osada J. 2013. Dietary oleanolic
acid mediates circadian clock gene expression in liver independently of diet and animal model but requires apoliproprotein
A1. J. Nutrit. Biochem. 21, 2100–2109. http://dx.doi.org/10.1016/j.jnutbio.2013.07.010 |
| ○ |
Guinda A, Albi T, Lanzón A. Procedure for the obtaining of oleanolic acid from the Olea europaea leaf. Spanish Patent 2001/2 160 553
|
| ○ |
Guinda A, Rada M, Delgado T, Gutiérrez-Adánez P, Castellano JM. 2010. Pentacyclic triterpenoids from olive fruit and leaf.
J. Agric. Food Chem. 58, 9685–9691. http://dx.doi.org/10.1021/jf102039t |
| ○ |
Hou W, Li Y, Zhang Q, Wei X, Peng A, Chen L, Wei Y. 2009. Triterpene acids isolated from Lagerstroemia speciose leaves as
α-glucosidase inhibitors. Phytotherapy Res. 23, 614–618. http://dx.doi.org/10.1002/ptr.2661 |
| ○ |
Kang W, Song S, Gu X. 2012. α-Glucosidase inhibitory in vitro and antidiabetic activity in vivo of Osmanthus fragans. J. Med. Plant Res. 6, 2850–2856.
|
| ○ |
Kang WY, Song YL, Zhang L. 2011. Alpha-glucosidase inhibitory and antioxidant properties and antidiabetic activity of Hypericum ascyron L. Med. Chem. Res. 20, 809–816. http://dx.doi.org/10.1007/s00044-010-9391-5 |
| ○ |
Khathi A, Serumula MR, Myburg RB, Van Heerden FR, Musabayane T. 2013. Effects of Sizigium aromaticum-derived triterpenes on
postprandial blood glucose in streptozotocin-induced diabetic rats following carbohydrate challenge. PLOS ONE 8, e81632. http://dx.doi.org/10.1371/journal.pone.0081632 |
| ○ |
Komaki E, Yamaguchi S, Maru I, Kinoshita M, Kakehi K, Ohta Y, Tsukada Y. 2003. Identification of anti-α-amylase components
from olive leaf extracts. Food Sci. Technol. Res. 9, 35–39. http://dx.doi.org/10.3136/fstr.9.35 |
| ○ |
Lacovicz JR. 2006. Principles of fluorescence spectroscopy (3rd edition). Springer. New York. |
| ○ |
Liu J. 2005. Oleanolic acid and ursolic acid: Research perspectives. J. Ethnopharmacology 100, 92–94. http://dx.doi.org/10.1016/j.jep.2005.05.024 |
| ○ |
Lowell BB, Shulman GI. 2005. Mitochondrial dysfunction and type 2 diabetes. Science 307, 384–387. http://dx.doi.org/10.1126/science.1104343 |
| ○ |
Minich DM, Bland JS, Katke J, Darlang G, Hall A, Lerman RH, Lamb J, Carroll B, Trypp M. 2007. Clinical safety and efficacy
of NG440: a novel combination of rho iso-alpha acids from hops, rosemary, and oleanolic acid for inflammatory conditions.
Can. J. Physiol. Pharmacol. 85, 872–883. http://dx.doi.org/10.1139/Y07-055 |
| ○ |
Prior RL, Hoang H, Gu L, Wu X, Bacchiocca M, Howard L, Hampsch-Woodill M, Huang D, Ou B, Jacob R. 2003. Assay for hydrophilic
and lipophilic antioxidant capacity (oxygen radical absorbance capacity (ORACFL)) of plasma and other biological and food
samples. J. Agric. Food Chem. 51, 3273–3279. http://dx.doi.org/10.1021/jf0262256 |
| ○ |
Quagliaro L, Piconi L, Assalone R, Martinelli L, Motz E, Ceriello A. 2003. Intermittent high glucose enhances apoptosis related
to oxidative stress in human umbilical vein endothelial cells. Diabetes 52, 2795–2804. http://dx.doi.org/10.2337/diabetes.52.11.2795 |
| ○ |
Rada M, Castellano JM, Perona J, Guinda A. 2015. GC-FID determination and pharmacokinetic studies of oleanolic acid in human
serum. Biomed. Chromatog. 29, 1687–1692. http://dx.doi.org/10.1002/bmc.3480 |
| ○ |
Re R, Pellegrini N, Proteggente A, Pannala A, Yang M, Rice-Evans C. 1999. Antioxidant activity applying and improved ABTS
radical cation decolorization assay. Free Rad. Biol. Med. 26, 1231–1237. http://dx.doi.org/10.1016/S0891-5849(98)00315-3 |
| ○ |
Schiekofer S, Andrassy M, Chen J, Rudofsky G, Schneider J, Wendt T, Stefan N, Humpert P, Fritche A, Stumvoll M, Schleicher
E, Häring H-U, Nawroth PP, Bierhaus A. 2003. Acute hyperglycemia causes intracellular formation of CML and activation of ras,
p42/44 MAPK, and nuclear factor κB in PBMCs. Diabetes 52, 621–633. http://dx.doi.org/10.2337/diabetes.52.3.621 |
| ○ |
Sharma O, Bhat TK. 2009. DPPH antioxidant assay revisited. Food Chem. 13, 1202–1205. http://dx.doi.org/10.1016/j.foodchem.2008.08.008 |
| ○ |
Song M, Hang TJ, Wang Y, Jiang L, Wu XL, Zhang Z, Shen J, Zhang Y. 2006. Determination of oleanolic acid in human plasma and
study of its pharmacokinetics in Chinese healthy male volunteers by HPLC tandem mass spectrometry. J. Pharmaceut. Biomed. Anal. 40, 190–196. http://dx.doi.org/10.1016/j.jpba.2005.06.034 |
| ○ |
Wang X, Li Y-L, Wu H, Liu J-Z, Hu J-X, Liao N, Peng J, Cao P-P, Liang X, Hai C-X. 2011. Antidiabetic effect of oleanolic acid:
A promising use of a traditional pharmacological agent. Phytother. Res. 25, 1031–1040. http://dx.doi.org/10.1002/ptr.3385 |
| ○ |
Wang X, Ye X-L, Liu R, Chen H-L, Bai H, Liang X, Zhang X-D, Wang Z, Li W-L, Hai C-X. 2010. Antioxidant activities of oleanolic
acid in vitro: Possible role of Nrf2 and MAP kinases. Chemico-Biological Interactions 184, 328–337. http://dx.doi.org/10.1016/j.cbi.2010.01.034 |
| ○ |
Watson JD. 2014. Type 2 diabetes as a redox disease. The Lancet 383, 841–843. http://dx.doi.org/10.1016/S0140-6736(13)62365-X |
| ○ |
Xu LZ, Wan ZX. 1980. The effect of oleanolic acid on acute hepatitis (70 cases). Human Medicine 7, 50–52.
|
| ○ |
Yan J, Zhang G, Pan J, Wang Y. 2014. α-Glucosidase inhibition by luteolin: Kinetic, interactions and molecular docking. Int. J. Biol. Macromol. 64, 213–223. http://dx.doi.org/10.1016/j.ijbiomac.2013.12.007 |
| ○ |
Yang ZG, Li HR, Wang LY, Li YH, Lu SG, Wen XF, Wang J, Daikonoya A, Itanaka S. 2007. Triterpenoids from Hippophae rhamnoides L. and their nitric oxide production-inhibitory and DPPH radical-scavenging activities. Chem. Pharm. Bull. 55, 15–18. http://dx.doi.org/10.1248/cpb.55.15 |
| ○ |
Yin M-C, Chan K-C. 2007. Nonenzymatic antioxidative and antiglycative effects of oleanolic acid and ursolic acid. J. Agric. Food Chem. 55, 7177–7181. http://dx.doi.org/10.1021/jf071242m |